
美国FDA发布ECMO设备紧急使用授权政策
应对新冠疫情呼吸治疗设备短缺,中国相关企业出口迎来机遇
为缓解新冠疫情期间呼吸治疗设备短缺问题,2020年4月6日,美国FDA发布了针对体外膜肺氧合(ECMO)及呼吸旁路设备的紧急使用授权(EUA),为中国ECMO设备生产企业出口美国提供了便利。
一、政策生效时限
该EUA仅在卫生与公共服务部(HHS)宣布的与COVID-19相关的公共卫生紧急事件期间有效。
二、适用范围
涵盖以下两类体外循环设备及其附件:
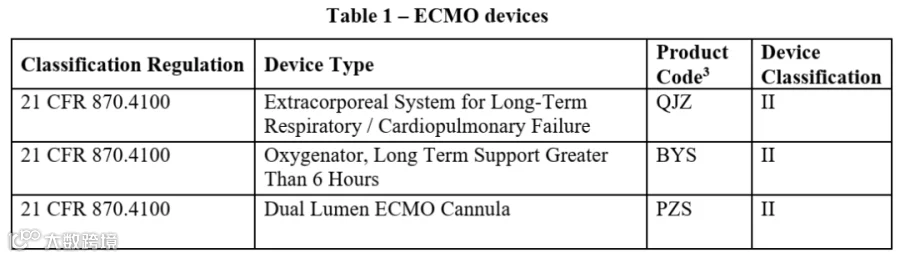
① 用于长期心肺衰竭治疗的ECMO设备(分类代码21CFR870.4100),可提供超过6小时的体外循环与血液气体交换;
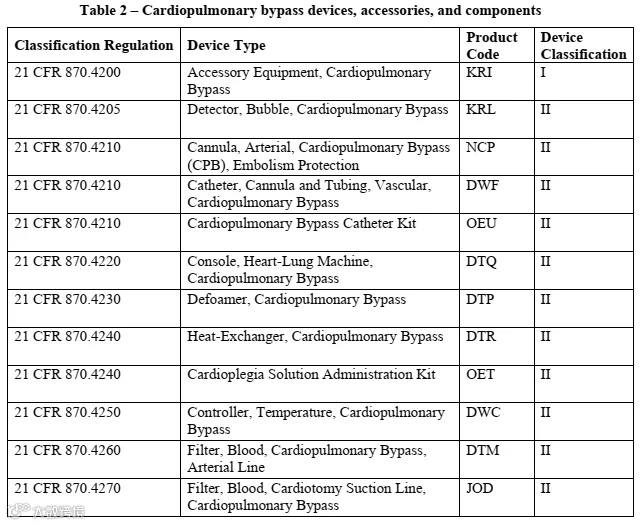
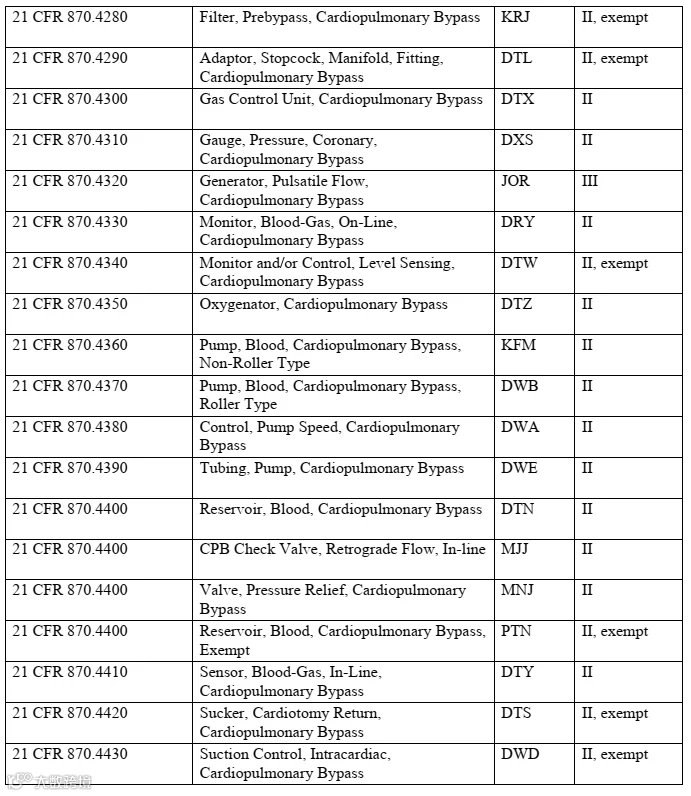
② 其他受21CFR870E多项法规管制、使用时间≤6小时的心血管手术体外循环设备。
上述设备需具备以下功能之一:输送血液至氧合组件、控制泵速、调节或监控气体流量、控制血液温度。
三、对已批准设备适应症的修改要求
FDA允许在不引发不当风险的前提下,对已获批设备的适应症或设计进行有限调整。
不视为不当风险的修改包括:
① 将体外循环设备用于急性呼吸/心肺衰竭患者的ECMO治疗;
② 延长设备使用时间超过6小时;
③ 更换不影响整体血流的回路附件(如套管、管道、过滤器等)。

可能产生不当风险的修改:
① 改变设备涂层;
② 影响气体交换性能的变更,例如:
a. 气体交换纤维尺寸变化;
b. 纤维或膜材料类型改变;
c. 纤维表面积调整。

修改后标签须包含:
① 新适应症或ECMO相关设计的数据说明,涵盖设备性能、耐久性、动物或临床表现及潜在风险;
② 明确提示设备更换必要性的临床观察报告;
③ 使用条件信息,明确区分已批准与未批准用途,并说明未经许可的变更内容。
FDA建议依据其认可标准开展设计、评估和验证工作。

四、申请EUA需提交的信息
尚未在美国销售但拟用于ECMO且支持超6小时体外循环的制造商,应向FDA提交以下资料:
① 企业基本信息及美国代理人联系方式;
② 产品标签复印件;
③ 是否已在其他地区获证(如CE、TGA、加拿大卫生部、日本厚生劳动省);
④ 设备性能数据;
⑤ 是否按照FDA认可标准完成设计与验证;
⑥ 生产是否符合21 CFR Part 820或ISO 13485质量体系要求;
⑦ 电源是否适配美国电压、频率及插头标准,或配备相应转换器。
以上信息需发送至CDRH-NonDiagnostic ETemplates@f-da.hhs.gov。若无法提供全部材料,可通过pre-EUA程序与FDA沟通。

五、授权使用条件
1. 确保医护人员知情:
① FDA已批准紧急使用;
② 已知与潜在的显著受益与风险;
③ 可用替代方案及其风险收益对比。
2. 确保使用者知情:
① 同上信息告知;
② 明确接受或拒绝使用的选项及后果。
3. 建立不良事件监测与报告机制,遵循21 CFR Part 803相关规定。
4. 制造商须满足记录保存与FDA访问审查的要求。

供稿单位:深圳海关
