
代谢相关脂肪性肝病(MASLD)已成为全球最普遍的慢性肝病,全球发病率高达 30%,严重时可进展为肝硬化甚至肝癌。其中,遗传因素在发病中扮演关键角色,TM6SF2 E167K 变异就是明确的风险因子,但它如何驱动肝脏脂肪堆积和损伤,一直是未解之谜。

青岛大学附属医院团队在《Clinical and Molecular Hepatology》(IF=16.9,JCR Q1 分区)发表重磅研究DOI: 10.3350/cmh.2024. 0268,通过基因敲入小鼠模型和脂质组学技术,首次揭示核心机制:TM6SF2 E167K 变异通过增强与 PNPLA3 的相互作用,阻断多不饱和脂肪酸(PUFA)从甘油三酯(TG)向磷脂酰胆碱(PC)的转移,最终引发肝脏脂肪变性和损伤。更重要的是,饮食补充含 C18:3 的 PC 可有效逆转这一病理过程,为 MASLD 精准治疗提供了新方向。
一、研究设计:层层递进解析致病机制
研究团队采用 “基因模型 - 机制解析 - 临床验证 - 干预验证” 的完整研究思路,逻辑清晰且证据链扎实:
构建 TM6SF2 E167K 基因敲入(KI)小鼠模型,喂食高脂饮食(HFD)构建 MASLD 模型,对比野生型(WT)小鼠的肝脏表型差异;
通过脂质组学分析肝脏脂质组成变化,锁定 PUFA-TG 和 PUFA-PC 的代谢失衡;
借助 Co-IP、免疫荧光等技术,验证 TM6SF2 与 PNPLA3 的相互作用及对 PUFA 转移的影响;
检测 MASLD 患者血浆脂质水平,明确 PC(16:0/18:3)的临床诊断价值;
在 KI 小鼠中进行含 C18:3 的 PC 饮食干预,验证治疗潜力。
二、核心发现:四大突破解码 MASLD 致病谜题
1.TM6SF2 E167K 变异显著加重肝脏脂肪变性和损伤
Fig. 1 通过体内外多层级实验,直接证实该变异的致病性,为后续机制研究奠定表型基础。
动物模型表型验证:高脂饮食(HFD)干预 16 周后,TM6SF2 E167K 基因敲入(KI)小鼠的体重、肝脏重量及肝指数均显著高于野生型(WT)小鼠(Fig. 1F-I)。HE 染色和油红 O 染色清晰显示,KI 小鼠肝细胞内脂肪空泡密集分布,脂肪变性程度远超 WT 小鼠(Fig. 1J),肝脏甘油三酯(TG)含量较 WT 小鼠升高 30% 以上(Fig. 1K)。
肝脏损伤量化证据:KI 小鼠的 MASLD 活动评分显著升高,血浆丙氨酸转氨酶(ALT)、天冬氨酸转氨酶(AST)水平分别较 WT 小鼠增加 68% 和 59%(Fig. 1M-N),提示肝细胞损伤严重,肝功能受损明显。
原代肝细胞功能验证:分离 KI 与 WT 小鼠原代肝细胞,经游离脂肪酸(FFAs)刺激后,KI 组肝细胞油红 O 染色吸光度值较 WT 组升高 98%(补充 Fig. 2),排除系统性因素干扰,证实变异肝细胞自身脂质蓄积能力显著增强。
TM6SF2 E167K 变异通过增强肝脏对脂质应激的敏感性,加速脂肪堆积与肝细胞损伤,是 MASLD 的关键遗传风险因子。
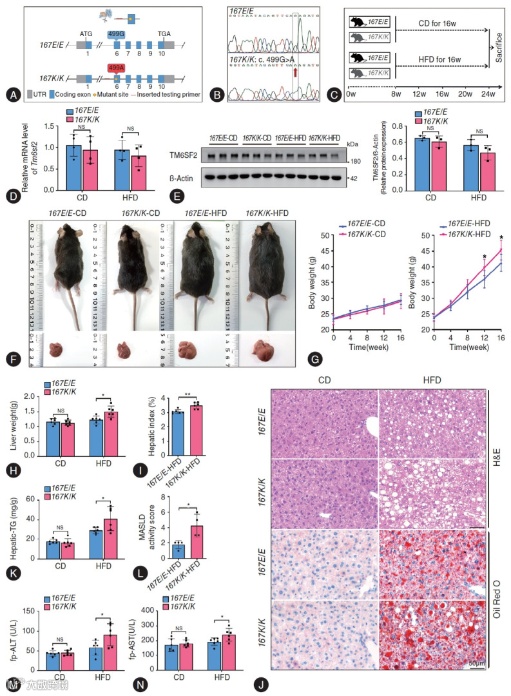
Fig. 1 TM6SF2 E167K变异体加重了小鼠高脂蛋白引起的肝脂肪变和损伤
2.脂质组学锁定核心代谢失衡 ——PUFA-TG 升高,PUFA-PC 降低
Fig. 2 通过非靶向脂质组学分析,从 1231 个脂质分子中精准定位变异导致的特异性代谢紊乱。
脂质总量失衡特征:KI 小鼠肝脏 TG 总量较 WT 小鼠升高 33.1%,而磷脂酰胆碱(PC)总量降低 25.6%(Fig. 2C-D),两者呈现 “一升一降” 的反向变化,打破肝脏脂质代谢平衡。
PUFA 亚型特异性紊乱:按脂肪酸饱和度分类后,核心失衡聚焦于多不饱和脂质(PUFA):KI 小鼠肝脏 PUFA-TG 含量较 WT 小鼠升高 41.5%,而 PUFA-PC 含量降低 42.4%(Fig. 2E-F),饱和脂肪酸(SFA)和单不饱和脂肪酸(MUFA)修饰的脂质无显著差异,证实变异导致的代谢紊乱具有亚型特异性。
转运平衡比值预警:KI 小鼠肝脏 PUFA-PC/PUFA-TG 比值仅为 0.36,显著低于 WT 小鼠的 0.88(Fig. 2G),提示 PUFA 从 TG 向 PC 的转运过程受阻,这是脂质堆积的核心代谢症结。
PUFA 在 TG 与 PC 之间的转运障碍,而非单纯脂质合成或分解异常,是变异致 MASLD 的关键代谢机制。
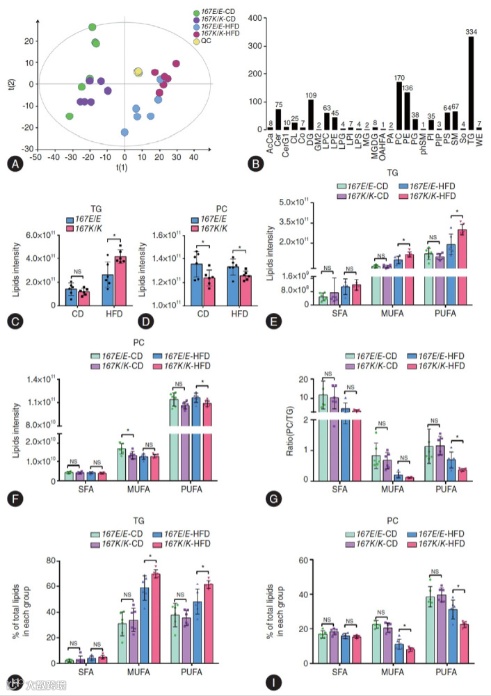
Fig. 2 TM6SF2 E167K变异体会导致高脂饮食小鼠肝组织中含PUFA的TG积累,并下调含PUFA的PC蛋白
3.机制揭秘 ——TM6SF2 与 PNPLA3 相互作用阻断 PUFA 转移
Fig. 3 和 Fig. 4 从分子互作与功能验证层面,共同阐明 PUFA 转运障碍的核心分子通路。
(1)蛋白相互作用增强与定位异常(Fig. 3)
免疫共沉淀(Co-IP)实验证实,TM6SF2 与 PNPLA3 存在直接相互作用,而 E167K 变异使两者结合强度较 WT 小鼠增强 89%(Fig. 3B)。
免疫荧光染色显示,WT 小鼠中 PNPLA3 主要定位于脂滴(LDs),而 KI 小鼠中 PNPLA3 被变异 TM6SF2 “滞留” 于内质网(ER),脂滴中分布显著减少(Fig. 3D-E、H-I),导致其无法在功能位点发挥作用。

Fig. 3 TM6SF2 E167K变异体增强了TM6SF2与PNPLA3蛋白之间的相互作用。
(2)PUFA 转运功能直接验证(Fig. 4)
采用炔基标记的亚油酸(PUFA 类似物)进行示踪,KI 小鼠肝细胞中新合成的 PC/TG 比值较 WT 小鼠降低 62%(Fig. 4C-D),直接证实 PUFA 从 TG 向 PC 的转运受阻。
敲低 PNPLA3 后,KI 与 WT 小鼠肝细胞的 PUFA-PC/PUFA-TG 比值差异消失(Fig. 4H-I),反向验证 PNPLA3 是介导该转运过程的关键蛋白,变异通过抑制其功能导致代谢紊乱。
这一突破破解核心机制:TM6SF2 E167K 变异增强与 PNPLA3 的相互作用,导致 PNPLA3 定位异常,阻断 PUFA 从 TG 向 PC 的转移,是 MASLD 发生的分子核心。
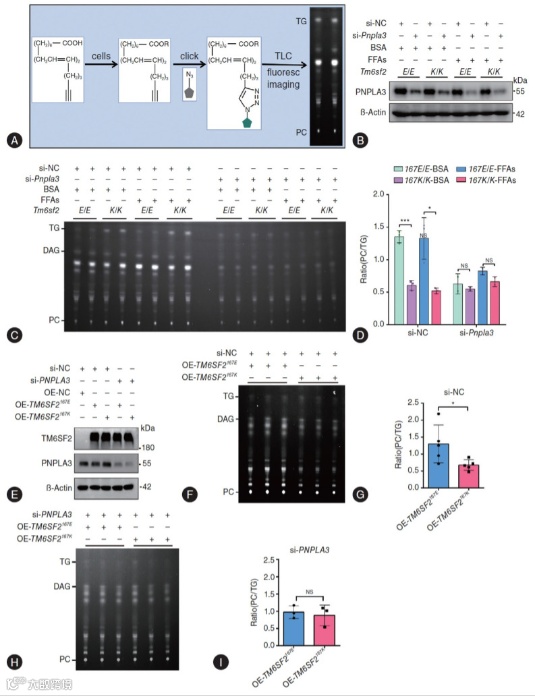
Fig. 4 TM6SF2 E167K变异体会损害PNPLA3介导的PUFA从TG转移到PC
4.氧化应激与临床转化 ——PC(16:0/18:3)既是标志物也是干预靶点
(1)氧化应激加剧损伤(Fig. 5)
KI 小鼠肝细胞经 FFAs 刺激后,活性氧(ROS)和脂质过氧化产物(MDA)水平分别较 WT 小鼠升高 72% 和 63%(Fig. 5A-B),细胞膜流动性降低 33%(Fig. 5F),透射电镜显示内质网结构断裂、碎片化(Fig. 5H),证实 PUFA 代谢失衡通过诱发氧化应激加重肝细胞损伤。
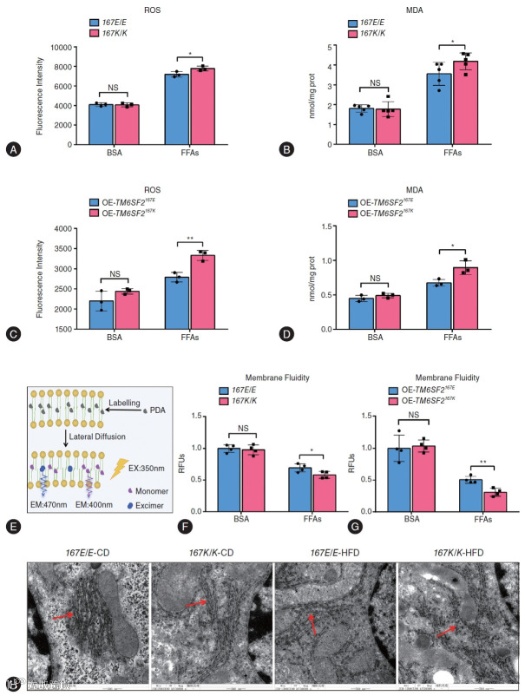
Fig. 5 TM6SF2 E167K变异体会导致脂肪酸处理肝细胞的MDA和ROS水平升高,并降低细胞膜流动性
(2)临床标志物鉴定(Fig. 6)
对 21 例 MASLD 患者和 11 例健康人血浆进行靶向脂质组学分析,携带 TM6SF2 E167K 变异的患者,血浆 PC(16:0/18:3)水平较非携带者降低 41%,且与肝脏脂肪含量呈显著负相关(r=-0.7095,P<0.05)(Fig. 6F-G)。该指标区分 MASLD 患者与健康人的 AUC 达 0.7446,区分变异携带者与非携带者的 AUC 为 0.7636(Fig. 6I-J),具备潜在临床诊断价值。

Fig. 6 携带TM6SF2 E167K变异的MASLD患者血浆中PC(16:0/18:3)减少
(3)饮食干预有效性验证(Fig. 7)
给 KI 小鼠补充含 C18:3 的 PC 后,肝脏 TG 含量较对照组降低 25%,ROS 和 MDA 水平分别降低 47% 和 43%(Fig. 7F、I),血浆 ALT、AST 水平显著回落(Fig. 7K-L),肝细胞细胞膜流动性恢复至接近 WT 小鼠水平(Fig. 7G),HE 染色显示脂肪变性程度明显减轻(Fig. 7H),成功逆转变异诱导的肝脏病理变化。
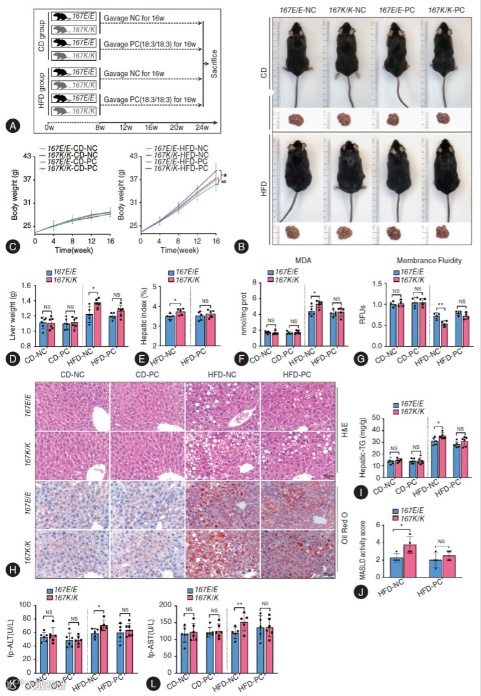
Fig. 7 含有C18:3的PC饮食干预显著减轻了高脂饮食小鼠中由TM6SF2 E167K诱导的肝脂变和损伤
这一突破实现 “诊断 - 治疗” 闭环:PC(16:0/18:3)可作为变异相关 MASLD 的临床标志物,而含 C18:3 的 PC 饮食补充则为精准干预提供可行方案。
三、核心创新点:三大维度填补研究空白
首次阐明 TM6SF2-PNPLA3 相互作用的致病角色:突破以往 “功能缺失” 的单一认知,揭示变异通过 “蛋白相互作用增强 - 酶功能阻断” 的新机制,为遗传因素致 MASLD 提供全新解释;
锁定 PUFA 代谢失衡的核心地位:明确 PUFA 从 TG 向 PC 转移受阻是关键病理环节,而非单纯的脂质合成或分解异常;
发现精准干预靶点:同时鉴定出 PC(16:0/18:3)作为临床标志物,且含 C18:3 的 PC 可作为饮食补充剂,实现 “诊断 - 治疗” 一体化,转化价值突出。
四、临床转化价值:从基因检测到精准干预
风险筛查:对 MASLD 高危人群进行 TM6SF2 E167K 基因检测,结合血浆 PC(16:0/18:3)水平,可早期识别高风险患者;
精准干预:针对变异携带者,饮食补充含 C18:3 的 PC(如富含该成分的植物油、营养补充剂),为 MASLD 提供个性化治疗方案;
诊断辅助:血浆 PC(16:0/18:3)可作为无创生物标志物,辅助 MASLD 的诊断和病情评估。
五、结语
该研究通过严谨的实验设计,首次完整揭示了 TM6SF2 E167K 变异驱动 MASLD 的分子机制,即 “增强 TM6SF2-PNPLA3 相互作用→阻断 PUFA 转移→PUFA-TG 积累 + PUFA-PC 减少→氧化应激→肝脏脂肪变性和损伤”。同时,明确了含 C18:3 的 PC 的诊断和治疗双重价值,为 MASLD 的精准防治提供了科学依据。
随着对 MASLD 遗传机制的深入解析,未来有望实现 “基因分型 - 标志物检测 - 个性化干预” 的全链条管理,让脂肪性肝病的治疗从 “广谱干预” 走向 “精准靶向”。
原文链接:https://e-cmh.org/journal/view.php?doi=10.3350/cmh. 2024.0268