
阿姆斯特丹医学中心Carol Ann Remme课题组在Cardiovascular Research杂志上发表了题为 “Chronically elevated branched chain amino acid levels are pro-arrhythmic” 的研究论文。作者发现Bcat2p.Q300*/p.Q300*小鼠由于BCAAs(支链氨基酸)分解代谢不完全,血浆支链氨基酸(BCAAs -亮氨酸、异亮氨酸、缬氨酸)水平急剧升高,体外诱发心律失常、心脏传导和复极障碍。作者的研究结果首次确定了BCAA升高与心律失常之间的因果关系,这暗示了BCAA代谢失调条件(如糖尿病、代谢综合征和心力衰竭)下心律失常发生的原因。

心律失常通常发生在有潜在病理的情况下,包括心肌缺血、结构紊乱。此外,患有代谢紊乱(糖尿病、肥胖)和心力衰竭的患者发生心律失常的风险增加。在N-乙基-N-亚硝基脲(ENU)诱变筛选中,作者发现了一个突变小鼠系,表现为突然死亡和Bcat2基因的纯合无义突变。受影响的小鼠血浆支链氨基酸水平急剧升高,原因是BCAA分解代谢不完全、心律失常易感性增强、心脏传导和复极障碍、离体心肌细胞内钙离子过度失调。在普通人群中,血浆BCAA水平与心电图(ECG)传导和复极指数呈正相关,这支持了BCAA在心电功能调节中的作用。由于对疾病机制和途径的认识不充分,妨碍了有效预防和治疗战略的发展。
作者的目的是用一种方法来确定潜在的心律失常和心源性猝死(SCD)的新机制。
纯合Bcat2p.Q300*/p.Q300*小鼠具有猝死表型且BCAA水平升高
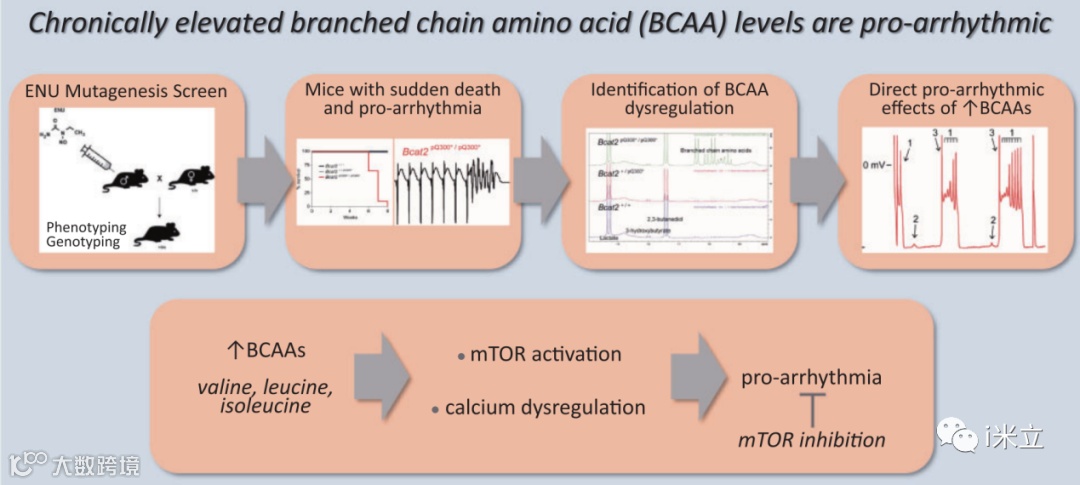
对C57BL/6J雄性小鼠(G0)进行ENU诱变和C3H连续育种。Pde6b+雌性(图1A), G3组诱变动物作为哈维尔老化筛查的一部分进行纵向表型研究。由混合背景G3小鼠组成的单一系谱小鼠在6-9周龄之间突然死亡,无明显原因,且无震颤或步态异常等前期症状(图1B)。杂合子Bcat2C1121T和野生型小鼠在8周龄前均无猝死,纯合小鼠在8周龄后均无存活(图2A)。Bcat2C1121T突变导致早期合成停止(Q300*),并因此导致BCAT2蛋白缺失CXXC基序。纯合子Bcat2p.Q300*/ p.Q300*小鼠的血浆中钾含量增加,但氯离子浓度降低,铁含量增加。虽然意义重大,但这些数据都没有为猝死表型提供一个明确的解释。
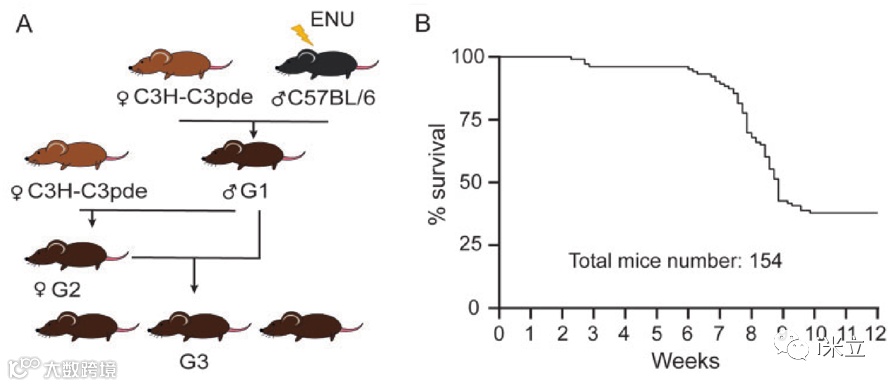
图1:ENU诱变诱导幼龄小鼠猝死表型
BCAT2负责分解BCAAs,在分解代谢为各自的a-酮酸(BCKA)后,BCAAs被支链酮酸脱氢酶复合物(BCKDC)进一步分解,最终的分解产物(乙酰辅酶a和琥珀酰辅酶a)最终在线粒体中被消耗,通过三羧酸循环进行呼吸。血浆核磁共振分析显示Bcat2p.Q300*/ p.Q300*5周龄小鼠血浆中BCAAs水平增加了10倍(但BCKAs没有增加,低于检测阈值)。杂合子Bcat2+/p.Q300*小鼠在6-8月龄前未出现猝死或其他明显表型,BCAA血浆水平正常(图2B-D)。因此,该突变导致BCAT蛋白的缺失,并导致BCAAs的积累,而不是BCKAs的积累。
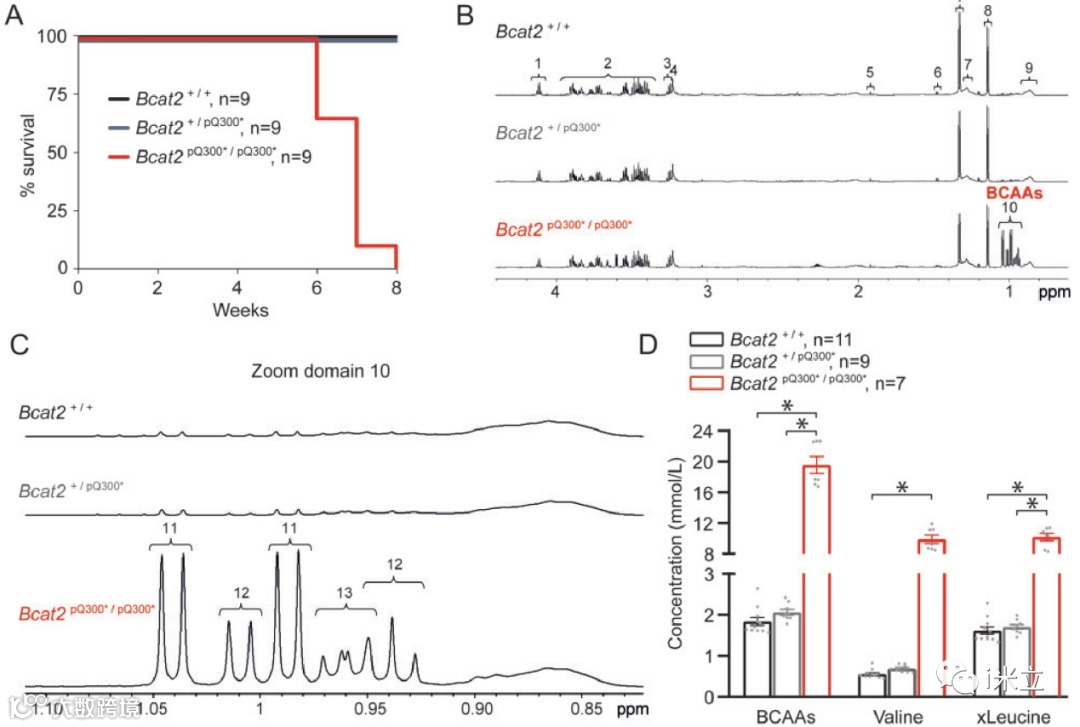
图2:纯合子Bcat2p.Q300*/ p.Q300*小鼠在年轻时出现猝死和血浆BCAA水平升高
Bcat2p.Q300*/p.Q300*小鼠显示心电改变和诱发性心律失常增加
作者对4-5周大的小鼠进行了详细的电生理分析,远早于6周大的小鼠突然开始死亡。异氟醚麻醉下表间心电图显示Bcat2 p.Q300*/ p.Q300*的QT间期、QTc间期和PR间期明显延长 (图3A-B)。接下来,作者研究了Bcat2+/+与Bcat2 p.Q300*/ p.Q300*之间的体外心脏电生理差异。Bcat2 p.Q300*/ p.Q300*小鼠房室传导时间与复极时间明显延长(图3B-F),提示APD前心律失常异质性增加。在灌注心脏中诱发性心律失常的研究显示Bcat2 p.Q300*/ p.Q300*中诱发性心律失常的发生率增加,其中大部分为非持续性(图3G-H)。综上所述,纯合Bcat2 p.Q300*/ p.Q300*小鼠体内心电图异常,体外房室传导时间、复极时间延长并显示出诱发型心律失常。

图3:纯合Bcat2 p.Q300*/ p.Q300*小鼠体内心电图异常,体外诱发型心律失常
Bcat2p.Q300*/p.Q300*心肌细胞动作电位延长、钙调节异常和前心律失常
图4A显示从Bcat2+/+和Bcat2p.Q300*/ p.Q300*分离的离体左心室心肌细胞以2Hz起搏频率诱发的典型AP和最大上搏速度(Vmax)。Bcat2p.Q300*/ p.Q300*心肌细胞的Vmax、APA和AP持续时间(APD90)均显著升高(图4B)。采用快速起搏(20次5Hz脉冲)方案,评估是否存在促心律失常事件,包括EADs、DADs和TAPs(图4C中箭头所示)。在Bcat2p.Q300*/ p.Q300*心肌细胞中,TAPs和EADs的发生率显著升高(图4D)。
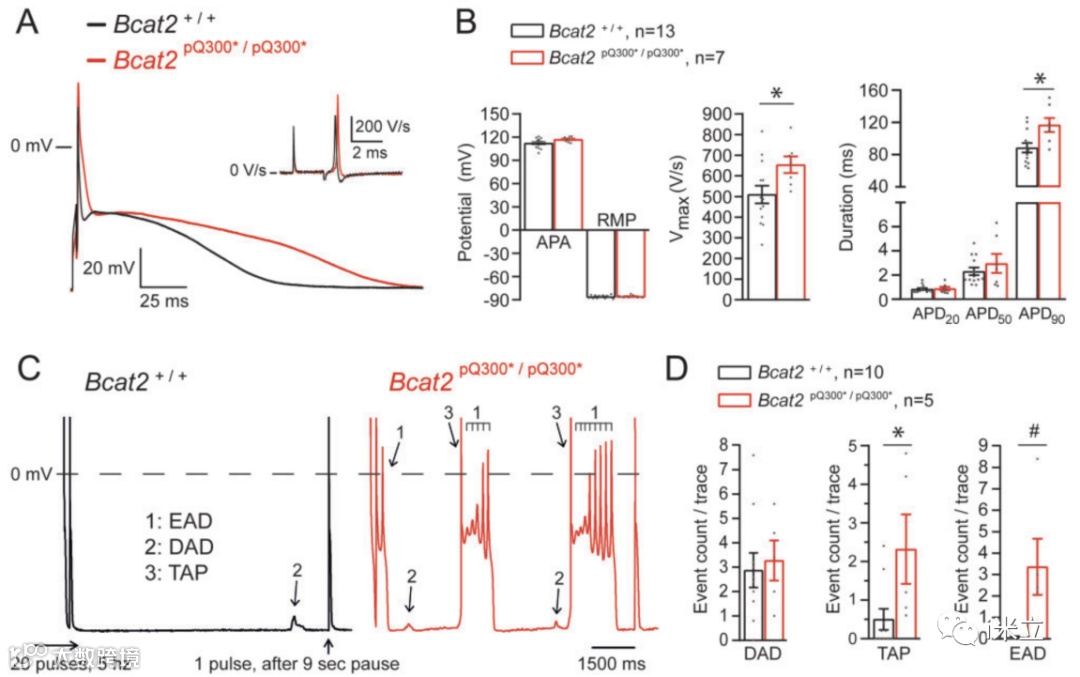
图4:Bcat2p.Q300*/p.Q300*心肌细胞前心律失常
图5A显示6Hz起搏频率下典型的钙浓度([Ca2+]i)瞬变。平均而言,Bcat2p.Q300*/ p.Q300*心肌细胞表现为细胞内舒张期钙离子水平增加([Ca2+]i)和瞬时钙离子浓度增加([Ca2+]i)(图5B)。Bcat2p.Q300*/ p.Q300*心肌细胞瞬时钙离子的总数量(触发和非触发联合)显著高于Bcat2+/+心肌 (图5C-D),表明钙依赖的促心律失常机制。
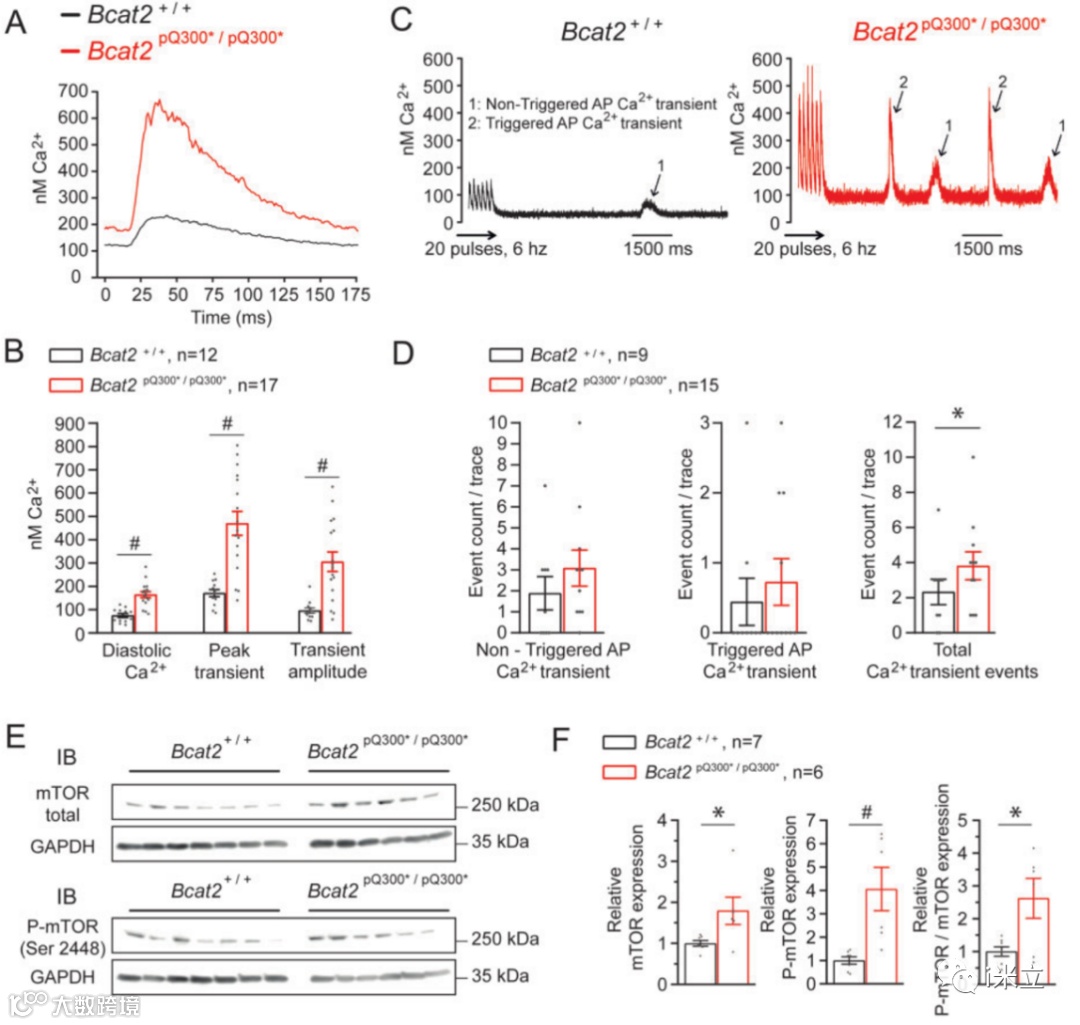
图5:Bcat2p.Q300*/p.Q300*小鼠细胞内钙异常,mTOR上调
BCAAs在人PSC衍生的心肌细胞中直接促心律失常
hPSC-CMs孵育5天,BCAA水平与Bcat2 p.Q300*/ p.Q300*血浆中BCAA水平相似,导致APD90显著延长(图6A-B),与在对照培养基中培养的hPSC-CMs相比,促心率失常早期和延迟后去极化(EAD/DAD)的发生率显著增加(图6C-D)。类似于Bcat2 p.Q300*/ p.Q300*的心肌细胞,在BCAA浓度增加的培养基中培养hPSC-CMs也表现出细胞内钙调节异常,包括舒张期([Ca2+]i)和瞬变后的钙调节异常(图7A-D),进一步证实了BCAAs直接导致心律失常的作用。

图6:BCAAs水平的增加再现hPSC-CMs的前心律失常表型
BCAA水平升高的促心律失常作用是由mTOR通路激活介导的
升高的BCAA浓度已被证明会导致mTOR活性的慢性诱导和氧化应激的增加。实际上,来自Bcat2 p.Q300*/ p.Q300*的心脏组织,mTOR总蛋白和激活的P-mTOR表达水平显著升高,表明mTOR途径激活(图5E-F)。与mTOR抑制剂雷帕霉素一起培养可防止APD90延长和EAD/DAD的发生(图6A-D),以及BCAA水平升高引起的钙调节异常(图7A-D)。这些观察证明了mTOR通路激活参与BCAA水平升高的促心律失常作用。

图7:BCAAs升高导致hPSC-CMs细胞内钙调节异常在mTOR抑制下是可逆的结合p.Q300*/ p.Q300*小鼠和人类样本的数据,这些发现表明更高的血浆BCAA水平与心脏传导和复极化改变有关。
小结
作者确定BCAA代谢是一种心电功能的新调节器,也是心律失常和猝死的中介。这种将BCAA与促心律失常联系起来的新机制可能不仅与遗传性BCAT2缺乏症相关,也与获得性代谢紊乱有关,如糖尿病、肥胖和BCAA代谢受损的心力衰竭。由于细胞BCAA代谢可由BCAA血浆水平反映,后者可作为心律失常和SCD风险的循环预后生物标志物。此外,这些代谢机制是可以通过饮食干预等方式改变的,提供了新的预防和治疗目标,克服了传统的针对离子通道或转运体的抗心律失常方法的缺点。此外,作者首次证明了通过药物抑制mTOR途径可以消除BCAAs升高引起的促心律失常事件。
总的来说,作者的发现为预防糖尿病、肥胖和心力衰竭患者的电功能障碍和心律失常的治疗策略的新研究铺平了道路。