


脊柱侧弯是指脊柱的一个或数个节段向侧方弯曲伴有椎体旋转的三维脊柱畸形,这些弯曲会影响身体的整体平衡和对齐,还可能导致其他身体健康问题。国际脊柱侧弯研究学会(Scoliosis Research Society, SRS)将脊柱侧弯定义为:应用Cobb法测量站立正位X光像的脊柱侧方弯曲,如角度大于10度则为脊柱侧弯。
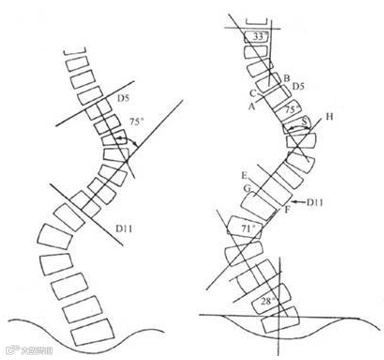
Cobb法
大部分脊柱侧弯的病因还不明确,多半认为是遗传因素及环境因素综合影响所造成。先天性脊柱侧弯是由近轴中胚层、体节或轴骨架的发育改变导致的。在脊椎动物胚胎发生中,近轴中胚层位于神经管附近,并在特定的过程中从胚胎的前部发展为体节,进而形成椎体,发育为完整的脊柱。这个过程主要由WNT、FGF和Notch信号通路调节,通路中的许多基因异常都会导致先天性脊柱侧弯的出现。

体节发生过程中的三个主要通路及其相关基因
脊柱肋骨发育不良(SCDO)是一类遗传机制相对比较明确的先天性脊柱侧弯疾病,多表现为椎体的多个节段性缺损,伴有肋骨异常,从而导致躯干较短(四肢长度正常);大部分呈非进行性轻度脊柱侧弯,偶有严重的脊柱侧弯;肋骨错位,融合的数目不固定,有时减少;胸廓的形状大致对称;新生儿的呼吸功能可能会因胸腔尺寸减小而受损,两岁时可能会改善到足以支持相对正常的生长和发育;肺功能严重受限的儿童出生时可能患有肺动脉高压;男性腹股沟疝的风险增加;可能会出现肾脏和心脏异常。
目前OMIM数据库已经收录6个亚型,多以常染色体隐性遗传为主,其中SCDO5相关基因TBX6基因的亚效等位基因常与该基因的致病突变共同作用导致疾病(SCDO5)发生,其中亚效等位基因可能为高频突变(亚洲人中为44%,欧洲人中为33%),这一研究突破了疾病的遗传学研究中占主导地位的“常见变异导致常见疾病”的理论框架,揭示了常见变异与罕见变异共同作用导致疾病发生的新机理。
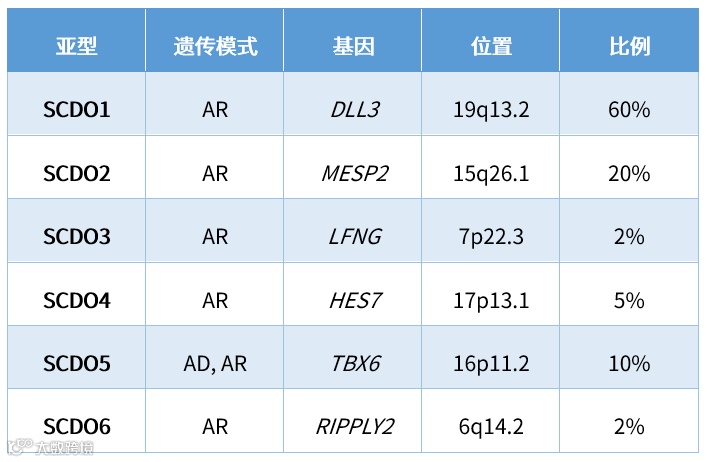
“脊柱肋骨发育不良”遗传学分型
TBX6基因位于16p11.2区域,是T-box家族的成员,编码参与发育过程调控的转录因子,在体节发生过程中发挥重要作用。目前已经明确TBX6基因的复合遗传机制与脊柱肋骨发育不良、脊柱侧弯相关。
2015年Wu等在The New England Journal of Medicine上发表文章“TBX6基因null变异联合常见亚效等位基因导致先天性脊柱侧凸(TBX6 Null Variants and a Common Hypomorphic Allele in Congenital Scoliosis)”,提出TBX6基因杂合致病突变不足以引起先天性脊柱侧弯,并确定了3个常见的TBX6基因TCA单倍型作为第二个风险等位基因,其中rs3809624和rs3809627位于5'UTR区域, rs2289292是8号外显子上的同义突变。他们提出了TBX6基因的复合遗传模型:先天性脊柱侧凸的罕见null变异(TBX6基因的无义、剪切、移码变异和16p11.2区域缺失)和反式位置常见风险等位基因的组合。该模型解释了文献中11%的中国先天性脊柱侧弯病例。

为了进一步明确TBX6的风险单倍型是功能性的,进行了连锁不平衡分析发现TBX6连锁不平衡区域很短,在该区域内并没有发现罕见的变异。同时还使用体外荧光素酶测定法来研究rs3809624和rs3809627对基因表达的影响,结果显示rs3809624和rs3809627上的非参考等位基因组合中TBX6基因的表达都有一定的削减。
 TBX6荧光素酶测定结果
TBX6荧光素酶测定结果
TBX6基因表达对于正常的椎骨形成至关重要。杂合的风险单倍型仅导致TBX6表达轻度降低,纯合子风险单倍型也不会显著降低TBX6表达。杂合的TBX6基因致病变异可能使基因表达减少一半。以上这些突变几乎不会达到先天性脊柱侧弯的基因剂量阈值。但是,TBX6的null变异和反式位置的风险单倍型会导致基因表达进一步下降,从而导致先天性脊柱侧凸的高风险。
这一研究突破了疾病的遗传学研究中占主导地位的“常见变异导致常见疾病”的理论框架,揭示了常见变异与罕见变异共同作用导致疾病发生的新机理。
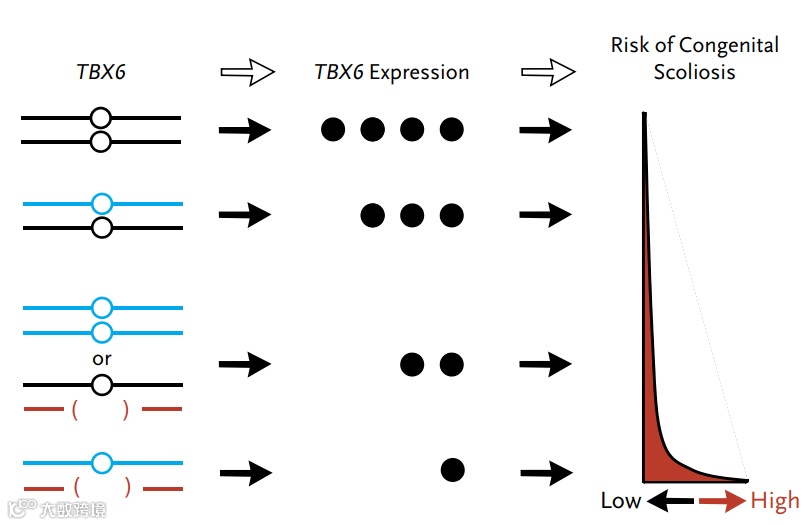
与TBX6相关的先天性脊柱侧凸的简化遗传模型
2019年,Liu等在Genetics in Medicine上发表文章“TBX6-associated congenital scoliosis (TACS) as a clinically distinguishable subtype of congenital scoliosis: further evidence supporting the compound inheritance and TBX6 gene dosage model”,通过分析TBX6相关的先天性脊柱侧弯患者可量化的表型,开发了临床诊断算法(TACScore)来辅助TBX6相关先天性脊柱侧弯的临床识别。
他们基于遗传学病因,将先天性脊柱侧凸分为TBX6基因相关性先天性脊柱侧凸(TBX6-associated congenital scoliosis,TACS)和非TBX6基因相关性先天性脊柱侧凸(Non-TACS),通过临床表型分析证实TACS患者临床症状出现更早(TACS,2y [1-3y];non-TACS,3y [1-9y]),以下半段脊柱的半椎体和蝴蝶椎为典型特征,且表现为更少的脊柱、肋骨和椎管内畸形受累。
 TBX6相关的脊柱侧弯:脊柱下部(T8–S5)受累更多
TBX6相关的脊柱侧弯:脊柱下部(T8–S5)受累更多
TACScore风险模型的变量包括(1)涉及下半部(T8-S5)的半椎/蝴蝶椎骨;(2)椎骨畸形的数量;(3)椎管内的存在缺陷;(4)肋骨缺陷的类型。该模型认为在TACScore大于或等于3的时候倾向于TBX6相关的先天性脊柱侧弯。

TBX6相关先天性脊柱侧弯的预测模型
2013年,Sparrow等曾报道过一个TBX6基因突变(c.1311A>T,p.*437Cext*81)引起的常染色体显性遗传家系,该变异位于终止密码子上,预计会导致在末端额外增加81个氨基酸,终止密码子及其上游的区域是高度进化保守的。体外的功能试验证明突变型TBX6蛋白(p.*437Cext*81)的转录激活活性约为野生型的50%,作者排除了显性负效应,认为是单倍剂量不足致病。非常遗憾的是文中没有对TCA风险单倍型进行相关分析。Liu等在介绍TBX6相关先天性脊柱侧弯的预测模型时指出,TCA单倍型在世界范围内很普遍(亚洲人中为44%,欧洲人中为33%),因此单倍型很可能会被测序数据的分析流程过滤掉。
在脊柱侧弯的遗传因素的探索过程中,了解TBX6基因的复合遗传机制对于数据分析和遗传咨询都是非常重要的。
脊柱肋骨发育不良遗传学分型延伸阅读
SCDO1型患儿在生命早期(胎儿或幼儿时期)每个椎体为圆形或卵形,边界光滑,这种现象被称为“卵石滩特征”。随着儿童期中期至晚期骨化的进行,卵石滩变为多个不规则形状的椎体和半椎骨。

SCDO2型患者所有椎骨节段均显示出一些形态和形状的异常;肋骨在肋骨交界处完全融合,从而产生“蟹状”特征;脊柱和躯干的缩短会严重影响儿童早期的呼吸功能。

SCDO3型患者脊椎的缩短比SCDO1和SCDO2型更严重,所有椎体均显示出更严重的畸形。
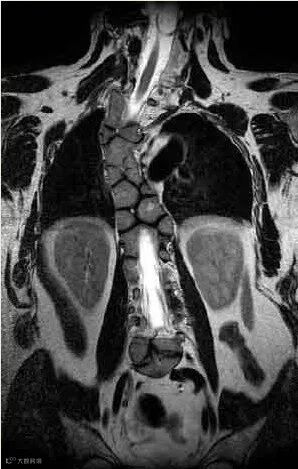
SCDO3型
❹ SCDO4型(HES7基因)
SCDO4型患者的所有椎体畸形程度都异常严重。与SCDO1相比,椎弓根相对突出(“蹦床”特征)。

SCDO4型
❺ SCDO5型(TBX6基因)
SCDO5型患者临床症状出现更早,以下半段脊柱的半椎体和蝴蝶椎为典型特征。

❻ SCDO6型(RIPPLY2基因)
SCDO6型患者多表现为颈椎椎骨缺失或融合等畸形,脖子短,运动受限。椎骨缺损会导致颈椎严重不稳定,在轻微的外部损伤后可能会导致心脏骤停。

SCDO6型:复杂的颅颈交接区畸形,C1-C3的后半部分缺失,C4和T9为左半椎,T4为右半椎
❼ DMRT2基因
已有文献报道DMRT2基因的纯合突变导致严重的肋骨畸形(缺失,融合和发育不良的肋骨),椎骨畸形(椎板椎间融合和椎体不规则骨化)以及轻度脊柱侧弯。患儿出生时存在严重的呼吸功能不全。DMRT2基因敲除的小鼠也表现出严重的肋骨和椎骨缺损。目前尚未被OMIM数据库收录。

严重的肋骨畸形,椎体的不规则骨化
往期回顾

