

1.BCAT2对于PDAC的发展至关重要
BCATs,包括BCAT1和BCAT2,将BCAA的氨基转移到α-酮戊二酸生成BCKA和谷氨酸(图1a)。随后,BCKA转化为支链酰基辅酶A酯,进而生成NADH。与对照组小鼠相比,KARS遗传基因突变的小鼠模型(KC小鼠)胰腺组织中6种必需氨基酸(包括BCAA)显著增加,而大部分非必需氨基酸没有变化(图1b)。PDAC细胞消耗的BCAA比正常人类胰管细胞多1.5-2.5倍(hTERT-HPNE,以下简称HPNE)(图1c)。与相邻细胞相比,BCAT1在腺泡、正常导管和PanIN(胰腺上皮内瘤)病变中的表达较低(图1d)。在KC小鼠PanIN导管细胞中观察到BCAT2高表达。总的来说,与相邻的正常导管细胞相比,PDAC细胞中BCAT2显著上调到更大程度。72份人类临床样本的PDAC导管细胞中,BCAT2普遍升高(图1e)。
将BCAT2敲除小鼠与KC小鼠杂交产生Pdx1-Cre;BCAT2flox/flox(以下简称Cre;BCAT2−/−)和LSL-KrasG12D;Pdx1-Cre;BCATflox/flox(以下简称KC;BCAT2−/−)小鼠(图1f)。在胰腺中敲除BCAT2可导致KC小鼠血浆中BCAA上调约3倍(图1g)。来自Cre;BCAT2−/−小鼠与表达BCAT2的小鼠的胰腺重量相比无差异,而与KC小鼠相比,KC;BCAT2−/−小鼠胰腺重量显著降低小鼠 (图1h)。BCAT2敲除显著降低了KC小鼠PanIN的进展(图1i)。与KC小鼠相比,KC;BCAT2−/−小鼠1、2、3期PanIN病灶从53.0%下降至31.5%,15.8%下降至6.5%,4.2%下降至检测不到 (图1j)。此外,PanIN病变的特征是酸性粘蛋白的大量产生和纤维化增强,这些效应在体内被BCAT2敲除所阻断 (图1k,l)。
总的来说,这些数据证明了BCAT2在PDAC发展中的重要作用。
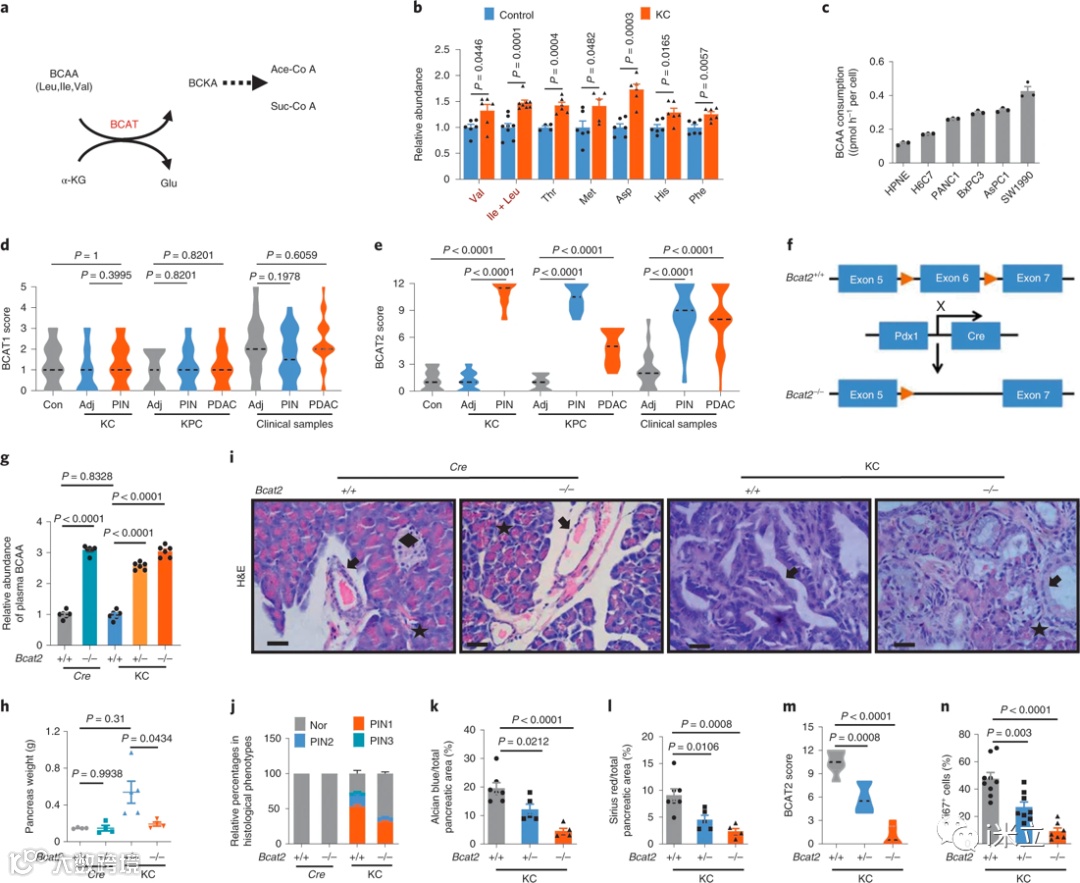
图1 BCAT2对于PDAC的发展至关重要
BCAT2的稳定过表达显著促进了细胞增殖,而BCAT2的下调则显著降低了细胞增殖(图2a-d)。在Panc02细胞中,多西环素(DOX)诱导的针对BCAT2 (shBcat2)的短发夹RNA (shRNA)不会影响肿瘤的形成,但显著降低了肿瘤的生长,并延长了小鼠的生存时间。给药BCAT2抑制剂或低BCAA饮食也显著延长了小鼠的生存时间(图2e)。BCAT2敲除降低了氧消耗率(OCR)并增加了细胞外酸化率(ECAR)(图2f-i)。此外,15N和13C亮氨酸示踪分析显示,BCAT2基因敲除降低了亮氨酸衍生的非必需氨基酸和α-酮异己酸生成(图2j-q)。在KC;BCAT2−/−产生的胰管类器官中观察到类似的结果。
KRAS过表达或敲低分别显著上调或下调BCAA代谢(图2r,s)。因此,PDAC细胞中过表达BCAT2支持线粒体呼吸并促进BCAA代谢。BCAT2抑制剂显著抑制KC胰管类器官生长;通过补充BCKA和碱基,这一效应得到了改善(图2t)。相反,BCAAs以剂量依赖的方式促进KC胰腺导管类器官的生长(图2u)。
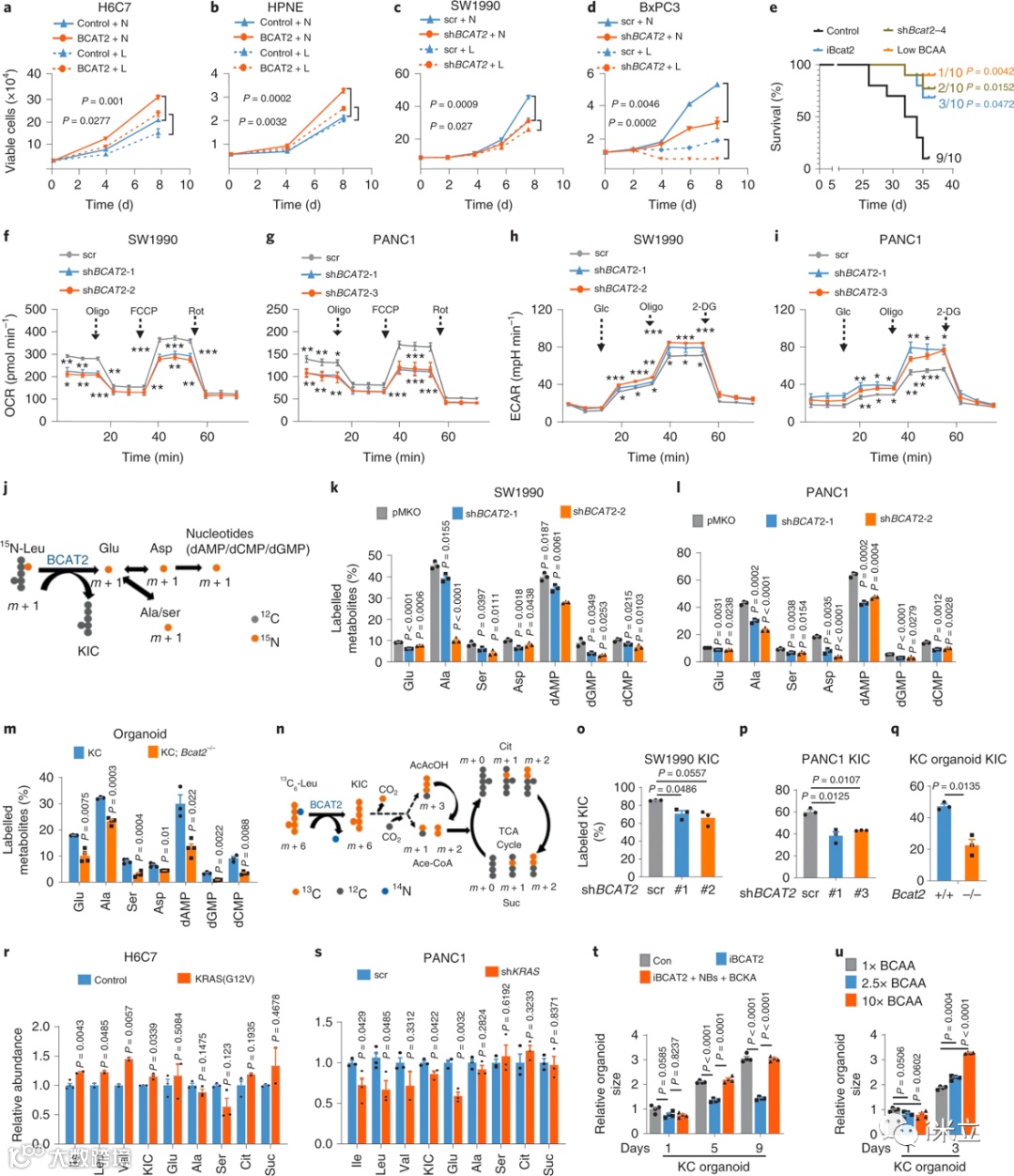
图2 BCAT2介导的BCAA分解代谢对PDAC至关重要
BCAT2过表达或下调BCAT2并不影响细胞内乙酰CoA水平和组蛋白乙酰化,这是通过抑制ATP柠檬酸裂解酶下调的(图3a-d)。此外,在KC小鼠中,BCAT2敲除后,腺泡细胞(而不是PanIN导管细胞)的组蛋白乙酰化显著降低(图3e),表明在腺泡-导管转化期间,腺泡细胞中BCAA衍生的乙酰辅酶A发挥重要作用,但在导管细胞中没有。

图3 BCAT2表达减少不影响胰管细胞组蛋白乙酰化
总的来说,这些结果表明,BCAT2上调通过增强线粒体功能维持了BCAA的利用以促进PDAC细胞恶性肿瘤进展,并在PanIN导管细胞中充当氮源。
3. KRAS抑制BCAT2的泛素化和降解
超过90%的人类PDAC发生KRAS突变,KRAS突变与BCAT2蛋白水平之间存在很强的正相关(图4a)。过表达KRASG12V显著上调BCAT2的表达,而敲除KRAS则降低了BCAT2的表达(图4b)。此外, KRAS过表达导致BCAT2泛素化程度降低,半衰期延长(图4c、d)。串联亲和纯化和质谱分析显示TRIM21是得分最高的BCAT2相互作用蛋白之一。KRAS基因敲除增强了BCAT2与内源性TRIM21的结合(图4e)。同样,TRIM21基因敲除增加了内源性BCAT2(图4f)。这些结果表明TRIM21靶向BCAT2进行降解。
在实验条件下,BCAT2的酪氨酸Y228可能是一个主要的磷酸化位点。为了确定Y228在BCAT2降解中的磷酸化机制,作者开始筛选具有激酶文库的潜在激酶,发现SYK是BCAT2降解的潜在候选激酶(图4g)。如图4h所示,SYK在体外激酶检测中直接磷酸化了BCAT2(WT)突变体,而不是BCAT2(Y228F)突变体。这些结果表明SYK是BCAT2在Y228位点的直接酪氨酸激酶。SYK敲除也增加了BCAT2水平(图4i)。此外,KRAS过表达降低了SYK的表达,但并未降低其磷酸化水平或激酶活性(图4j)。KRAS突变体样本中SYK的表达显著降低,而PDAC样本中BCAT2和SYK呈负相关关系(图4k-l)。因此,KRAS通过抑制Y228磷酸化来稳定BCAT2,从而减少TRIM21 E3连接酶介导的BCAT2降解(图4m)。
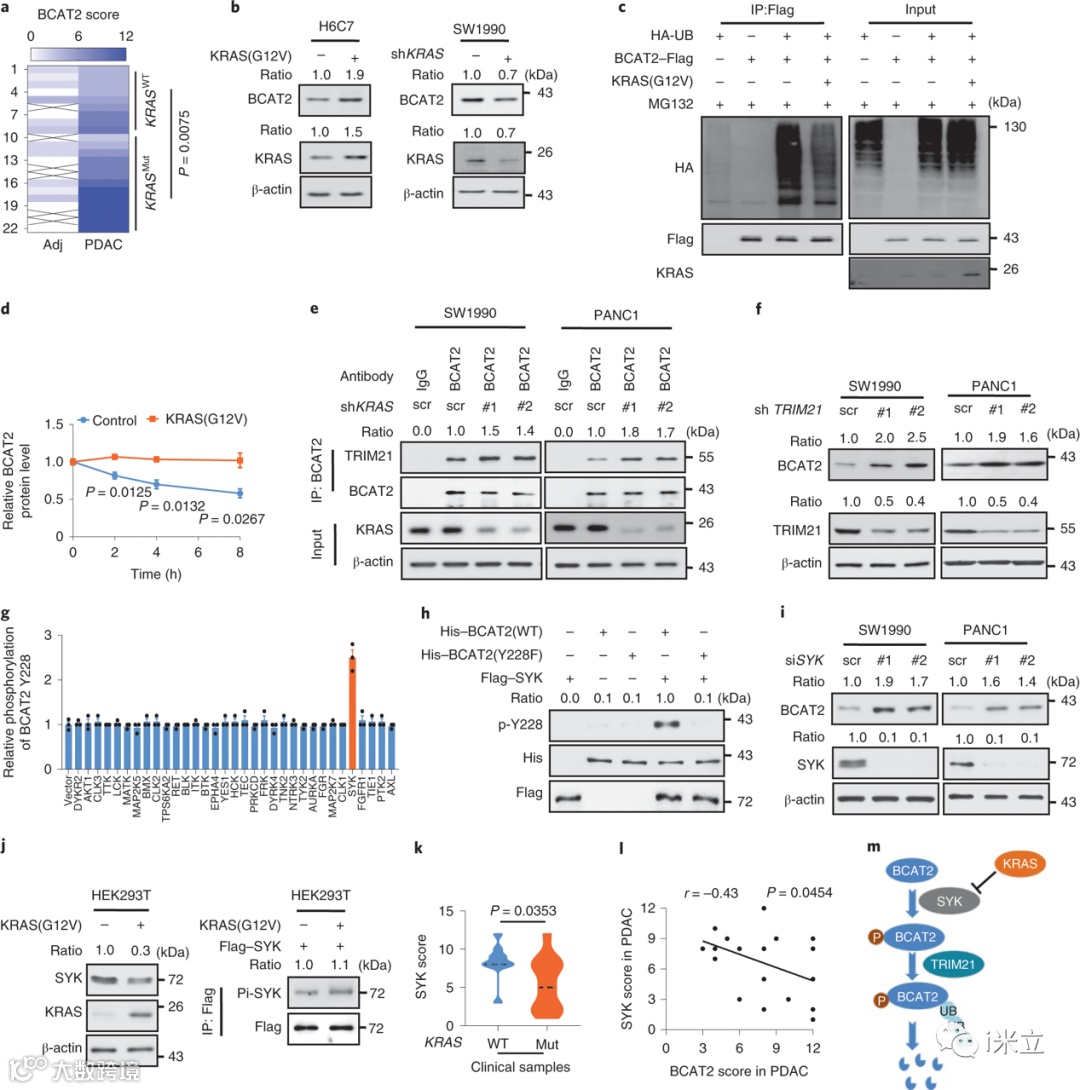
图4 KRAS抑制BCAT2的泛素化和降解
4. BCAT2抑制剂或低BCAA饮食会阻碍PDAC
给6个月大的KC小鼠注射BCAT2抑制剂两个月,并在8个月大时对其进行检测(图5a)。BCAT2抑制剂治疗5 d后,对照组和KC小鼠血浆BCAA浓度显著上调(图5b)。给药BCAT2抑制剂并没有改变对照组小鼠的胰腺重量(图5c,d),但导致KC小鼠胰腺重量显著降低(图5c,d)。此外,BCAT2抑制剂降低了KC小鼠PanIN病变的总负荷(图5e,f)。综上所述,体内功能抑制表明BCAT2在PDAC早期发育中具有重要作用。
作者使用定制的低BCAA饮食进行喂养,其中含有正常饮食中五分之一的BCAA。小鼠在1个月大时喂食这种低BCAA饮食,并在2个月、4个月和6个月时进行检测(图5g)。低BCAA饮食导致对照组和KC小鼠血浆BCAA浓度显著降低。KC小鼠的胰腺重量在4和6月龄时显著降低,KC小鼠和对照组的胰腺重量差异变得最小。此外,KC小鼠在2、4和6月龄时,低BCAA饮食显著阻碍了PanIN的发育(图5h,i)。用低BCAA饮食喂养1个月大的KPC(他莫昔芬诱导胰腺导管腺癌)小鼠两个月,并在3个月大时对其进行检测(图5j)。低BCAA饮食显著阻碍了KPC小鼠PDAC的发育和胰腺重量(图5k,l)。此外,低BCAA饮食显著降低了KPC小鼠的细胞增殖(图5m)。
因此,干扰BCAA-BCAT2轴阻碍了PDAC的发展。

图5 BCAT2抑制剂或低BCAA饮食会阻碍PDAC
作者揭示了BCAT2在小鼠模型和人类PDAC中升高。在LSL-KrasG12D/+;Pdx1-Cre小鼠中,胰腺组织特异性敲除BCAT2可阻碍PanIN的进展。在功能上,BCAT2促进BCAA摄取,以维持BCAA分解代谢和线粒体呼吸。值得注意的是,BCAA以剂量依赖的方式促进KC小鼠胰管类器官的生长。此外,KRAS可以稳定BCAT2, 该过程是由酪氨酸激酶(SYK)和E3连接酶蛋白21 (TRIM21)介导的。值得注意的是,在PDAC小鼠模型中,低BCAA饮食也会阻碍PDAC的发展。因此,BCAT2介导的BCAA分解代谢对含有KRAS突变的PDAC的发展至关重要。靶向BCAT2或降低膳食BCAA可能具有临床转化意义。
总之,作者揭示了BCAA - BCAT2轴在PDAC发育中的关键作用,以及PDAC中BCAA代谢重组的潜在机制(图5n)。
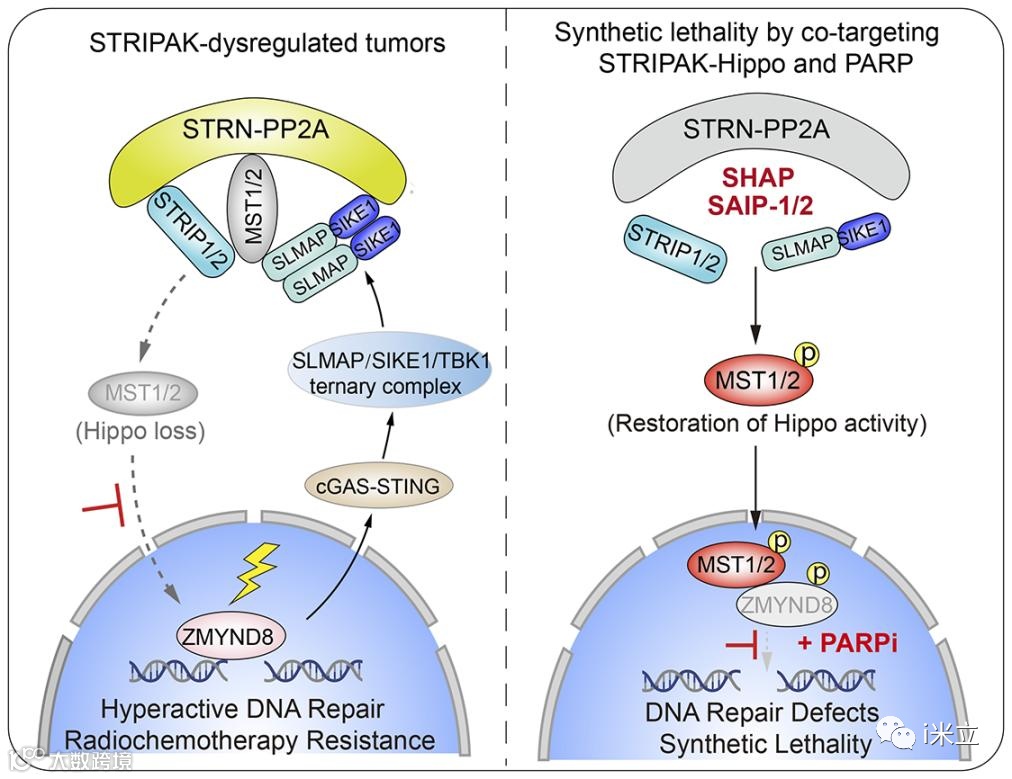
作者确定了Hippo-STRIPAK复合体在DNA双链断裂(DSB)修复和基因组稳定性的控制中是一个重要的角色。具体来说,总之发现哺乳动物的STE20样蛋白激酶1和2 (MST1/2),独立于经典的Hippo信号,直接磷酸化锌指MYND类型8 (ZMYND8),从而抑制细胞核中的DNA修复。为了应对遗传毒性应激,研究人员确定IFN基因的循环GMP-AMP合成酶/刺激因子(cGAS/STING)通路通过TANK连接激酶1诱导(TBK1诱导)IKBKE 1-肌膜相关蛋白抑制因子(SIKE1-SLMAP)臂的结构稳定,将核DNA损伤信号传递给Hippo-STRIPAK的动态组装。因此,作者发现STRIPAK2介导的MST1/2失活增加了癌细胞的DSB修复能力,并赋予这些细胞对放化疗和PARP抑制的抗性。重要的是,在动物和患者来源的肿瘤模型中,以三种不同的肽抑制剂中的每一种为靶点的STRIPAK组装有效地恢复了MST1/2的激酶活性,以抑制DNA修复,并使癌细胞对PARP抑制剂重新敏感。