
6月9日,CFDA集中发布了多项关于仿制药质量和疗效一致性评价的征求意见稿,包括《关于仿制药质量和疗效一致性评价工作有关事项的公告》、《仿制药质量和疗效一致性评价受理审查指南(需一致性评价品种)》和《仿制药质量和疗效一致性评价受理审查指南(境内共线生产并在欧美日上市品种)》,并且同时发布了《仿制药参比制剂目录(第五批)》和《仿制药参比制剂目录(第六批)》。CFDA在文件中明确了参比试剂的选择、生物等效性实验、欧美日上市的仿制药、一致性评价的申报和审批等多项流程。
图表1:仿制药质量和疗效一致性评价工作流程
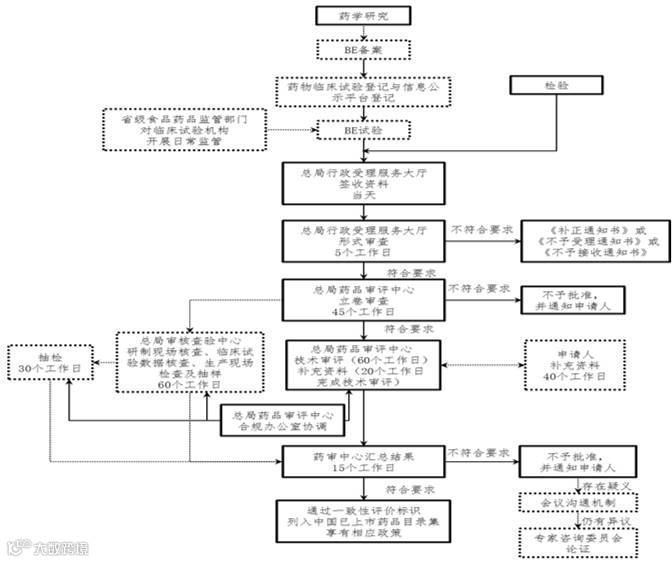
资料来源:CFDA、中航证券金融研究所整理
参比试剂选择方面,由CFDA列出289个品种的原研企业药品清单并向社会公布,供企业选择参比制剂时参考。企业自行采购参比试剂。
生物等效性实验(BE实验)实行备案管理,企业可以在现有经认定的临床试验机构进行,也可以在其他具备条件的机构进行。企业可以聘请具备评估能力的第三方按药物临床试验质量管理规范(GCP)要求对开展生物等效性试验的机构进行评估。药监局分批公布不适合开展人体内研究的药物品种。
药监局支持国内药企生产的在欧盟、美国和日本批准上市的药品在境内上市,包括已在境内上市使用同一生产线生产、已在境内上市使用不同生产线生产和未在中国上市的等各种情况,按照对应的实施规则执行。
一致性评价申报和审评流程同之前的预期一致,申报流程的确定依药品种类、生产和上市情况而定。申报获得受理后,CFDA药品审评中心对企业申报资料进行立卷审查。符合要求的,于45天内予以立卷。药品审评中心根据立卷审查情况提出有因检查和抽检的需求,有因检查工作一般在立卷审查结束后60天内完成。审评工作一般应当在受理后120天内完成。经审评认为需申请人补充资料的,申请人应在40天内一次性完成补充资料。审评工作完成后,CFDA会向社会公开审评结论。
通过明确实验资料、一致性评价申报流程和不同环节的时间节点,仿制药质量和疗效的一致性评价工作有望快速推进。从BE实验的时间需求和目前的时间窗口来看,相关企业的需求有望从17年下半年开始集中释放。
回归到市场,我们认为在既定的时间范畴内,仿制药质量和疗效一致性评价的工作进度可能会呈现前松后紧的状态,政府开展、执行政策的决心和力度维持不变。在相关流程确定后,企业需求的集中释放有望引爆CRO产业链,同时利好创新企业提前卡位、扩大市场份额。我们建议继续从两个维度分享政策红利,一是创新和制剂出口企业,包括华海药业、恒瑞医药等;另一方面,密切关注CRO行业的业绩兑现情况及产业链的龙头企业,包括泰格医药、博济医药等。