2024年3月22日,欧盟发布了24年度最新CECP概览。在此之前,欧盟于2023.1发布了首份CECP年度概览,包括了2021年4月-2022年6月提交的通知,有8个公告机构根据Article 54(3)提交了215份通知,有24(占比11.2%)台器械要求CECP,191台器械被豁免CECP。
CECP是MDR法规Article 54(1)中的临床评价咨询流程,由欧盟委员会专家小组执行。
MDR法规Article 54(4)(5)中规定如下:
委员会应起草一份符合附录IX第5.1节或附录X第6节流程的器械年度总结。该年度总结应包括本条第3段和附录IX第5.1节(e)点规定的公告决议以及公告机构未听从专家小组建议而被编入的器械列表。委员会应将该总结提交至欧洲议会、理事会和医疗器械协调小组。
委员会应2025年5月27日之前起草关于本条实施情况的报告,并将其提交欧洲议会和理事会。提交报告中应包括年度总结和MDCG提出的相关可用建议。根据本报告,委员会应酌情提出修订本法规的建议。
MDR法规Article 54(1)中规定如下:
适用于进行CECP的器械:
(a) III类可植入器械;和
(b) 附录VIII第6.4节(规则12)所述的用于给予和/或清除药品的IIb类有源器械。
本次的CECP概览,统计数据为2022年7月1日至2023年6月30日:
1.适用器械:
本次共有13家公告机构根据Article 54(3)发出了 353 份通知,根据Article 54(1)适用CECP程序的有36台器械(占比10.1%),317台器械根据Article 54(2)被豁免CECP。36个器械中有34个为III类植入式器械(94.4%),2个为IIb类ARMP器械(5.6%)。
大多数(58.4%)属于以下4大类:
a. 心脏功能植入式装置(16.7%);
b. 骨科假体(16.7%);
c. 听觉主动植入式装置(13.9%);
d. 植入式神经刺激器(11.1%)。


2.豁免器械:
在获得 CECP 豁免的317台器械中,285个为III类植入器械,32个为IIb类ARMP器械(10.1%)。大多数获得CECP豁免的器械(55.9%)属于以下三大类:
a.心血管植入物和人工心脏相关器械(12.0%)
b.骨科假体,骨合成器械,肌腱和韧带合成器(30.0%)
c.植入假体和骨合成器械-各种规格(13.9%)
d.6例未明确EMDN类型
在99.1%的器械(314个器械)中,豁免的原因是对同一制造商已经销售的、具有相同预期用途的器械进行了修改;其中2个器械,根据MDR (0.6%)(Article 54(2)(a))更新了证书;其中一个器械,公告机构声称豁免是由于通用规格可用(0.3%)(MDR Article 54(2)(c))。


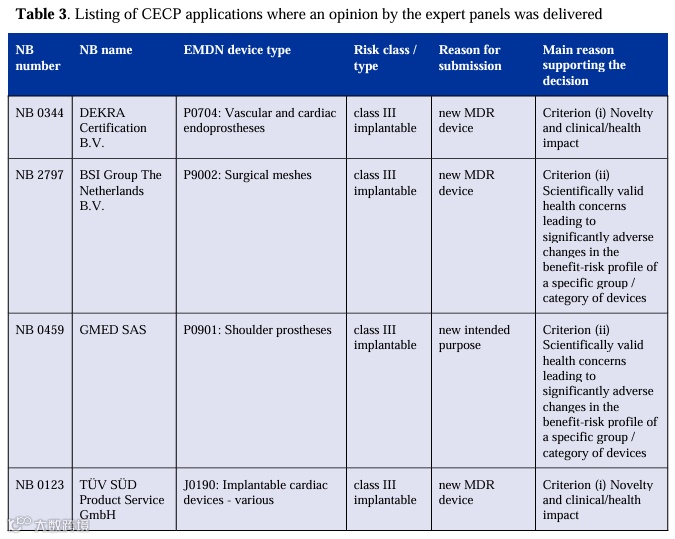
3.创新器械:
(i)器械的创新性或可能涉及和主要临床影响或健康影响的相关临床操作;
(ii)从特定品类器械或器械组受益风险比上观察产生的显著有害变化,是由有关组件或原材料,或有关器械故障的情况下对健康有影响的,科学有效的健康隐患所引起;