AlphaGenome:不依赖实验预测基因组及其变异对基因表达、剪切和分子互作的影响
写在前面的
前段时间DeepMind公司发布了继Alphafold3之后的又一生物学AI大模型:AlphaGenome。该模型旨在预测 基因组序列中突变对下游基因表达及其调控的分子生物学影响。AlphaGenome可以自定义分析基因组任意区域长达1Mb长度的DNA序列,特别是在非编码区(占人类基因组的 98%),并可提供预测区域内下游基因调控过程中分子间互作的site by site level的高分辨率景观。本期推送将初步分享关于该模型如何在本地搭建或网页使用方法。
AlphaGenome简介
解读基因组变异对下游基因表达调控的影响仍然是生物学领域的一大核心挑战。特别是在人类基因组中存在高达98%的非编码区。目前的研究发现非编码区的变异也能通过多种方式影响基因的转录、翻译甚至是蛋白的修饰。但如何大规模研究基因组变异对下游生物学过程的影响在基于传统分子生物学实验的策略下显得压力重重。特别是解读非编码变异如何调节基因组特性,例如染色质的可及性、表观遗传修饰和染色质的三维构象。如果没有计算预测,对绝大多数变异的复杂效应进行整体表征仍然困难重重。
针对上述问题,DeepMind公司开发了针对基因组及其变异对基因表达调控影响预测的AI大模型AlphaGenome,该模型属于一个将多模态预测、长序列上下文和碱基对分辨率统一到一个框架中的模型。该模型当前支持最长为100万碱基长度的DNA序列作为输入,并预测多种细胞类型的各种基因组轨迹。该团队利用一套全面的基准测试评估了AlphaGenome的预测性能,并证明了模型具备在先前未见过的DNA序列上准确预测基因组轨迹的能力,以及其在变异效应预测任务中的有效性。AlphaGenome在24个基因组轨迹预测任务和26个变异预测任务中取得了SOTA性能(SOTA, State of the Art,指代在特定任务和数据集上,当前最优秀且最先进的技术的性能表现,属于领跑者水平)。DeppMind公司对AlphaGenome在目标分辨率、序列长度、精炼和模态组合中进行了广泛的测试,以解释AlphaGenome的性能并为未来的序列到功能模型的设计选择提供参考。未来AlphaGenome或许可以为分析基因组内的调控代码提供一个强大且可扩展的基础。
如何搭建AlphaGenome分析平台
目前DeepMind团队公开了全部关于AlphaGenome的源代码在https://github.com/google-deepmind/alphagenome上。相比于Alphafold3,AlphaGenome的安装更简便。AlphaGenome可以直接通过pip安装,完成安装后申请私人API密钥即可使用。私钥申请网址https://deepmind.google.com/science/alphagenome/account/terms
conda create -p ~/gpu-software/miniconda3/envs/alphagenome python=3.10
conda activate ~/gpu-software/miniconda3/envs/alphagenome
git clone https://github.com/google-deepmind/alphagenome.git
pip install ./alphagenome
完成安装后即可使用AlphaGenome。
本地使用AlphaGenome预测突变对基因表达的影响
我选择人类基因组22号染色体的第35677410到36725986区间的序列用于示范如何编写python代码调用AlphaGenome预测突变对RNA转录的影响。我选择缺失人类基因组22号染色体第35687410位点处及其下游的600bp序列作为突变体序列,该分析的适用场景为基因编辑敲除某一段序列前评估是否缺失会影响基因的表达,或者在产前诊断中预测变异是否影响胎儿的基因表达。
#!/usr/bin/python
from alphagenome.data import genome
from alphagenome.data import gene_annotation, genome, track_data, transcript
from alphagenome.models import dna_client
from alphagenome.visualization import plot_components
import matplotlib.pyplot as plt
API_KEY = 'APIforUserDeep@Minds20250708'
model = dna_client.create(API_KEY)
#选择人类基因组22号染色体第35677410到36725986位置的序列进行研究
interval = genome.Interval(chromosome='chr22', start=35677410, end=36725986)
#构建参考序列的变体序列
variant = genome.Variant(
chromosome='chr22',
position=35687410,
reference_bases='N'*600,
alternate_bases='',
)
#构建模型预测
outputs = model.predict_variant(
interval=interval,
variant=variant,
ontology_terms=['UBERON:0001157'],
requested_outputs=[dna_client.OutputType.RNA_SEQ],
)
#绘制基因表达预测结果
plot_components.plot(
[
plot_components.OverlaidTracks(
tdata={
'REF': outputs.reference.rna_seq,
'ALT': outputs.alternate.rna_seq,
},
colors={'REF': 'dimgrey', 'ALT': 'red'},
),
],
interval=outputs.reference.rna_seq.interval.resize(2**15),
# Annotate the location of the variant as a vertical line.
annotations=[plot_components.VariantAnnotation([variant], alpha=0.8)],
)
plt.savefig('variant_rna_seq_plot.pdf', format='pdf', bbox_inches='tight')
plt.show()
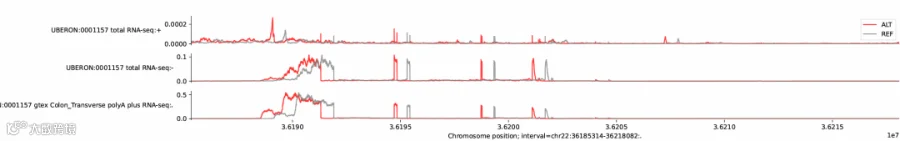
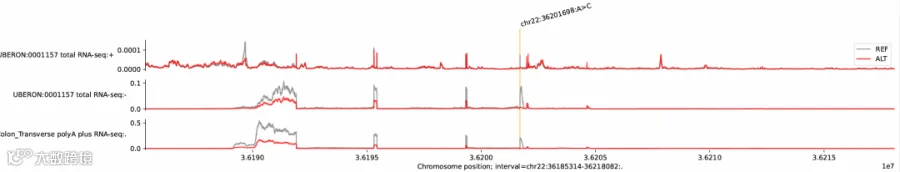
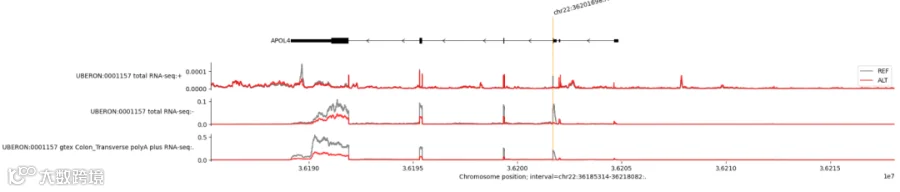
运行此代码可以得到名为variant_rna_seq_plot.pdf的文件,打开文件可看见图片中红色的线是突变体基因表达的水平,灰色的线是正常基因型的基因表达水平。由于本身缺失了600bp,红色的线会出现前移,但是从表达量可知,缺失这600bp对表达的影响较小。但是如果替换36201698位点的A碱基为C碱基,预测结果显示表达量显著降低。
variant = genome.Variant(
chromosome='chr22',
position=36201698,
reference_bases='A',
alternate_bases='C',
)
本地使用AlphaGenome预测序列在三维基因组水平的相互作用
作为示例还是选择刚才提到的那段序列以研究三维基因组水平下人类基因组中某特定片段与其他序列的相互作用
from alphagenome.data import genome
from alphagenome.data import gene_annotation, genome, track_data, transcript
from alphagenome.models import dna_client
from alphagenome.visualization import plot_components
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
API_KEY = 'APIforUserDeep@Minds20250708'
model = dna_client.create(API_KEY)
#构建序列对象
interval = genome.Interval(chromosome='chr22', start=35677410, end=36725986)
# Load gene annotations (from GENCODE).
gtf = pd.read_feather(
'https://storage.googleapis.com/alphagenome/reference/gencode/'
'hg38/gencode.v46.annotation.gtf.gz.feather'
)
# Filter to protein-coding genes and highly supported transcripts.
gtf_transcript = gene_annotation.filter_transcript_support_level(
gene_annotation.filter_protein_coding(gtf), ['1']
)
# Extractor for identifying transcripts in a region.
transcript_extractor = transcript.TranscriptExtractor(gtf_transcript)
# Also define an extractor that fetches only the longest transcript per gene.
gtf_longest_transcript = gene_annotation.filter_to_longest_transcript(
gtf_transcript
)
longest_transcript_extractor = transcript.TranscriptExtractor(
gtf_longest_transcript
)
longest_transcripts = longest_transcript_extractor.extract(interval)
ontology_terms = [
'EFO:0002824', # HCT116 colon carcinoma cell line.
]
#预测序列在三维基因组水平的相互作用
output = model.predict_interval(
interval=interval,
requested_outputs={dna_client.OutputType.CONTACT_MAPS},
ontology_terms=ontology_terms,
)
#绘制互作图
plot = plot_components.plot(
[
plot_components.TranscriptAnnotation(longest_transcripts),
plot_components.ContactMaps(
tdata=output.contact_maps,
ylabel_template='{biosample_name}\n{name}',
cmap='autumn_r',
vmax=1.0,
),
],
interval=interval,
title='Predicted contact maps',
)
plt.tight_layout()
plt.savefig('variant_contactmap_plot.pdf', format='pdf', bbox_inches='tight')
plt.show()
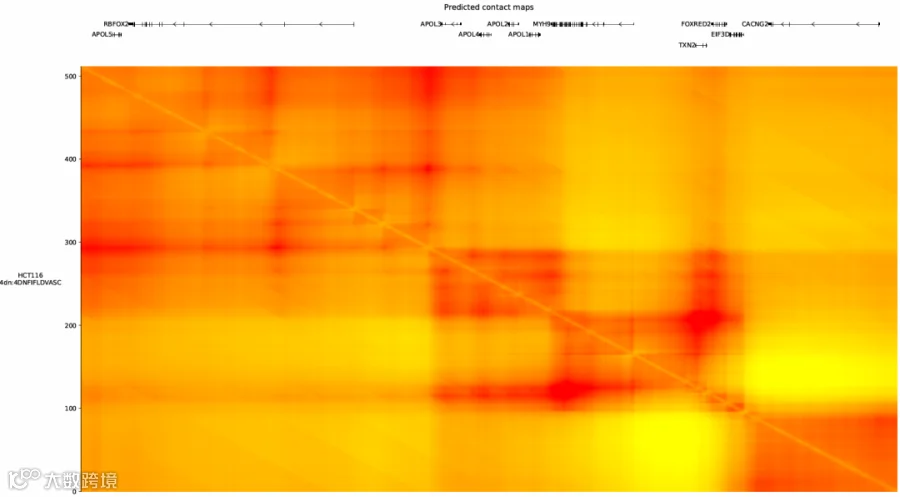
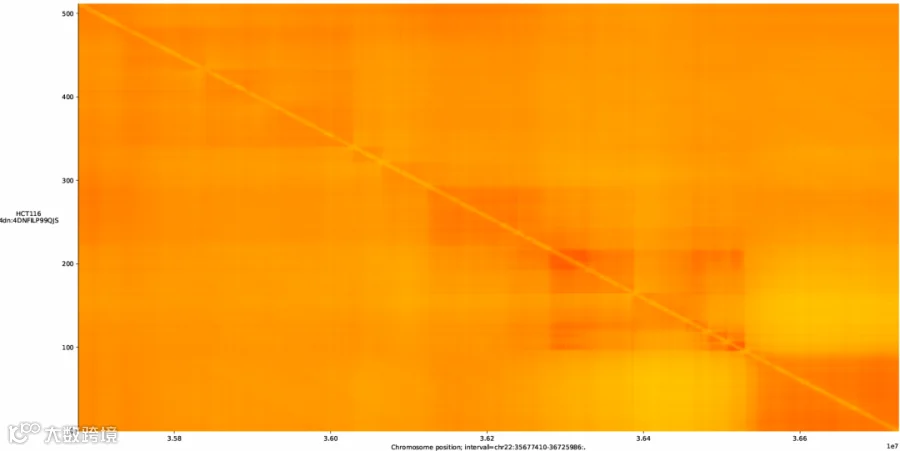
运行程序后可得到相互作用图,通过研究该图可以探究这段序列中哪些区域可能存在较强的相互作用。
利用Google Colab在线预测基因组变异对基因表达调控的影响
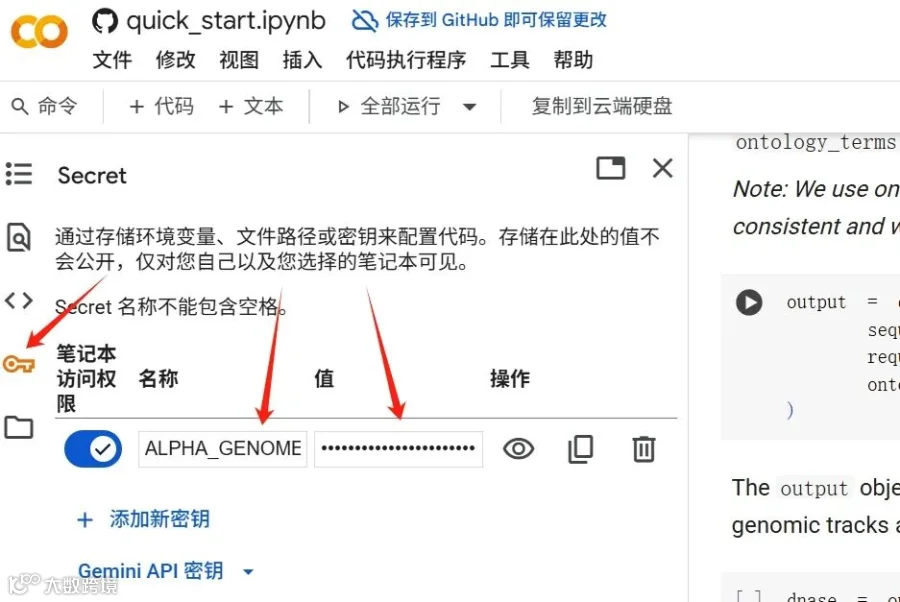
如果用户缺乏相应的编程背景,也可以使用Google Colab完成在线预测基因组变异对基因表达调控的影响,在线使用网站的好处在于简单方便。但是坏处在于无法批量运行分析大规模变异。比如上面的代码可以通过增设外部传参可以实现大规模批量分析。我在GPU的A100卡上完成上面两个程序的分析不到3分钟。在线使用AlphaGenome的网址为https://colab.research.google.com/github/google-deepmind/alphagenome/blob/main/colabs/quick_start.ipynb#scrollTo=U2Q-amDjkYVt进入网址后首先点击”钥匙“形状的按钮创建一个工作区,如下图箭头所示,工作区名称自定义,并输入密钥保存。完成保存后即可点击图片中的”全部运行按钮“实现在线预测基因组变异对下游生物学过程的影响。
AlphaGenome可靠性评价
阅读AlphaGenome原文时DeepMind团队提及通过了SOTA测试,虽然我手里没有实际的实验数据验证我做的这些分析的可靠性,但是AlphaGenome出自DeepMind团队之手,而DeepMind团队开发了目前全球广泛使用的Alphafold2和Alphafold3蛋白质结构预测及其与生物大分子相互作用的预测软件。我个人相信AlphaGenome必将在未来掀起基因组学、表观遗传学和医学领域的新一场浪潮。
写在后面的
AlphaGenome除了可以实现本期推送中提及的分析,还可以预测可变剪接、组蛋白修饰、以及利用ATACseq和RNAseq等多模态数据预测变异对基因表达、染色质可及性等十多种多维度生物学过程的分析。如果以后有机会,我会一一分享如何进行相关分析。
往期精彩
进阶(一):利用Alphafold3构建蛋白质结构宇宙——从PDB到私人数据库的范式跃迁
实战(三):Alphafold3批量实现蛋白与蛋白、蛋白与核酸、蛋白复合体与核酸相互作用预测