
如何一次性组装尽可能完整噬菌体基因组
写在前面的
上期推文主要讲述了如何根据基因组草图绘制估计噬菌体基因组大小。核心目标是要理解如何根据kmer总数和kmer深度计算估计基因组大小。同时,理解由于实验不可避免原因噬菌体基因组测序数据中会包含部分细菌基因组,这也是导致噬菌体基因组估计大小比实际基因组大的原因。
组装噬菌体基因组
基因组估计完后,就是对基因组的组装。而实际组装中有一个很有意思的现象。理论上说,测序深度越深,越有利于获得完整基因组。但是实际情况并不是这样。就拿我来说,之前我们研究所有其他实验室的师姐把她的噬菌体测序数据给我组装,她反馈给我的情况是使用spades组装,2G的测序大小组装效果还没有1G测序大小的组装效果好。她很不理解这是为什么。因为,她可以保证两次测序的实验准备都是很好的,建库也是很好很完整的。但是实际组装却是1G大小组装出了130kb,2G大小只组装出了96kb。这里测序批次问题并没有影响多大。(测序公司给出的回复)对于这个情况,我们来仔细分析一下。为什么2G的组装比1G的组装效果差。首先目前二代测序基因组De novo组装主流是基于De Bruijn graph进行组装。这是一种数学图论中的知识,如果接触过一笔画的同学可能会对这种方式有更深刻的认识。上篇推文中已经介绍了,在基因组分析中首先会将reads打散成更短的kmer,在基因组组装的过程中,这些kmer就派上了用场。reads被打断成长度为k的核酸片段(kmer)后,利用De Bruijn graph根据kmer间重复的部分构建graph,得到最优化路径从而拼接contig。那么如何利用kmer进行基因组组装呢?这里用网上的一幅图来助大家理解。两段reads AGATCTTGTTATT和GTTATTGATCTCC,选择长度为5的kmer打散。再根据kmer之间的overlap构建路径。如图从AGATC开始,依次往后步移一个碱基,当步移到红色方框的kmer时有了两种可能,由于两段reads有一段GTTATT的overlap,红色方框对应的kmer恰好有两种步移路径,一条是ATCTT(沿着第一条read的方向),一条是ATCTC(第二条read的方向),这两条路径最后都有不同的组装方式。对于第二条read, 绿色部分GTTAT->TTATT恰好又是它的起始也是第一条read的结束。那么根据下图的第一幅路径图可以优化为第二幅路径图。这样就将reads连接起来了。
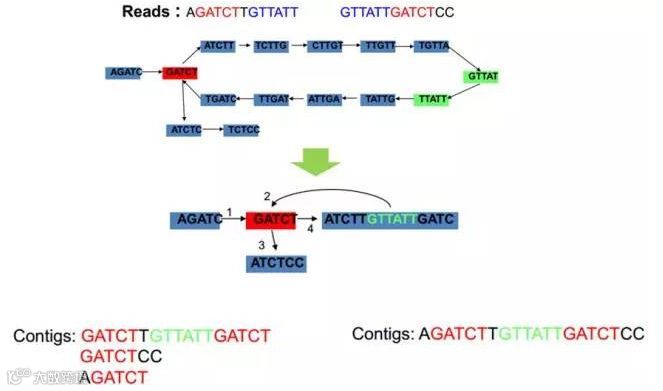
这么一看好像发现不了测序深度越深对基因组组装的影响。而事实上,我们忽略了重复序列对基因组装的影响,而且这个问题在二代测序中是暂时无法解决的。由于重复序列的存在,对于基因组的组装是有很大的影响。短序列在重复区域根本无法判断来自于哪个重复区段,程序在遇到重复序列时无法判断其来源,只能在这个部分分割为两段contigs,那么测序深度越深,在覆盖度相同的情况下重复区域被测序的次数增大,重复区域的reads数量增大。反而不利于实际的基因组组装。那么我们只能降低重复序列或者只取部分reads来组装了。只取部分reads可以保证重复序列数量减少,有利于组装软件组装更长的scaffolds。
今天我们先介绍只取部分reads组装。从fastq数据中取出部分reads进行组装,就要保证每条reads都有等可能的概率被抽取,这样随机性抽取才可以保证组装的合理性。那么这个问题就可以转化成从fastq数据中随机抽取reads了。这个问题,连明哥在他的BioLeetCode题目中就出过一个题目。在写程序前,我们要注意一个细节,抽取过程要保证双端fastq抽取随机相同的reads,一般来说下机后的fastq中reads都是配对的,但是部分reads过滤软件在trim低质量reads是会产生unpaired reads。(Trimmomatic不会,这个软件是trim掉一条质量低的序列会自动把另外一条序列丢进unpaired文件里)。所以我下面写的Perl程序默认是raw data操作。这样可以保证reads被随机抽取后一定是配对的。另外为了照顾对perl语言不太了解的小白,我没有使用任何perl或者bioperl的函数包,任何函数都是自己造车轮。
#!/usr/bin/perl -w
=head1 Name
Sub_readsFastq.pl
=head1 Description
Extract sub reads from PE fastq file.
Not: PE fastq file need raw or clean (trimomatic) fastq file by trim. (only contain paire reads)
=head2 Usage
perl Sub_readsFastq.pl total_fastq_lins percent_sub R1.fastq R2.fastq
=head2 Author
Dongyan Xiong
=head3 Edit time
2019-08-07 16:22 1.0
=cut
#编写寻找最大值的子程序,用于fastq的总序列数计算
sub max{
#子函数的参数传递用@_
my @arr = @_;
#接收参数传递进来的第一个值
my $max_num = $arr[0];
#利用循环判断最大值
for(my $i=0; $i<scalar(@arr); $i+=1,){
if($max_num <= $arr[$i]){
$max_num = $arr[$i];
}
else{
next;
}
}
#返回最大值
return $max_num;
}
#输入fastq文件的行数,可以在shell中用cat或者zcat统计行数
my $fastq_line = $ARGV[0];
#$per是percentage参数,输入你想提取的reads比例,0~1取值范围,即输入你想随机抽取百分之几的reads
my $per = $ARGV[1];
#统计fastq文件中有多少条序列
my $rand_num = int($fastq_line/4*$per);
#定义随机数哈希表
my %rand_hash;
#定义随机数数组
my @rand_hash;
#利用循环生成随机数把小于fastq文件的reads总数的随机数放入随机数数组中
while(@rand_hash < $rand_num){
#生成小于reads总数的随机数
my $num = int(rand($fastq_line/4));
#由于fastq文件一条reads有4行记录,我们只取reads第一行
$num = 4*$num-3;
#将以使用的随机数放入哈希的键中,只有不存在于哈希键中的随机数才可以被利用,保证reads不会重复被取到
push @rand_hash, $num."\n" if (!exists $rand_hash{$num});
$rand_hash{$num} = 1;
}
#对随机数列表进行排序,保证接下来取reads是从fastq文件从前往后抽取的
my @rand_hash_sort = sort { $a <=> $b } @rand_hash;
#由于上面采用了4*$num-3取fastq序列的第一行,有可能$num会产生0的随机数,这样就会有一个4*0-3的负数,必须要判断是否有负数,否则程序会报错
if($rand_hash_sort[0]<0){
#剔除数组第一个元素
shift @rand_hash_sort;
};
my $mx = max(@rand_hash_sort);
#定义将输出的fastq R1sub文件
my @out_fq1;
#打开fastq R1 raw data
open(FASTQ1, "<", $ARGV[2]);
my $i = 1;
my $k = 0;
#使用while循环逐行读取文件,保证内存消耗最小
while(my $id = <FASTQ1>){
my $res;
$i是定义来记录fastq文件行数的参数,由于我们生成的随机数对应的就是每个fastq文件中reads第一行在fastq文件中的行数,只要我们读取到fastq文件某一行,且这一行恰好在排序好的随机数数组里,那么我们就取走这条fastq序列(取走4行)
if($i."\n" eq $rand_hash_sort[$k]){
$k += 1;
$res = "TRUE";
}
else{
$res = "FALSE";
}
if($res eq "TRUE"){
push @out_fq1, $id;
my $sequence = <FASTQ1>;
push @out_fq1, $sequence;
my $comment = <FASTQ1>;
push @out_fq1, $comment;
my $quality = <FASTQ1>;
push @out_fq1, $quality;
#上面几行使得文件句柄对应的指针往后移动了几行,我们要将$i的值增加,用于保证$i的值读取文件的行数一致
$i+=4;
}
elsif($i > $mx){
#如果$i已经超过了fastq文件最大行数,就停止操作了
last;
}
else{
#否则继续操作
$i+=1;
}
}
#对于R2 fastq同样如R1那样操作
open(FASTQ2, "<", $ARGV[3]);
$i = 1;
$k = 0;
my @out_fq2;
while(my $id = <FASTQ2>){
my $res;
if($i."\n" eq $rand_hash_sort[$k]){
$k += 1;
$res = "TRUE";
}
else{
$res = "FALSE";
}
if($res eq "TRUE"){
push @out_fq2, $id;
my $sequence = <FASTQ2>;
push @out_fq2, $sequence;
my $comment = <FASTQ2>;
push @out_fq2, $comment;
my $quality = <FASTQ2>;
push @out_fq2, $quality;
$i+=4;
}
elsif($i > $mx){
last;
}
else{
$i+=1;
}
}
#写出R1
open(OUT1, ">>", "sub-R1.fastq");
print OUT1 @out_fq1;
#写出R2
open(OUT2, ">>", "sub-R2.fastq");
print OUT2 @out_fq2;
使用这个perl程序抽取随机抽取fastq序列,内存消耗小,速度也快。我师姐2G的数据,抽取50%只花了十几秒。用sub fastq文件(经过尝试只随机取走10%),放入SPAdes组装噬菌体,最后得到了138kb噬菌体基因组,总长度与全长完全一致!
本次重点
了解在实际组装噬菌体基因组中,并不是测序深度越深越好,重复序列数量的增加会严重降低组装效率!可以采用随机抽取部分序列进行组装。效果显著!另外,在编写这个perl脚本时有以下几个知识点
1.使用while逐行读取fastq数据不耗内存,是非常节省计算资源的方法;
2.由于使用了while逐行读取,而取fastq数据是一次取4行,会造成文件句柄指针往后移动,需要通过正确计算保证指针与参数匹配;
3.随机数生成要注意生成非冗余随机数,保证一条reads不会被重复抽取,保证抽取的随机性与完整性;
4.添加了自定义百分比,可以通过输入参数定义任意抽取比例。
作者信息
熊东彦,中国科学院武汉病毒研究所在读研究生。擅长转录组分析,宏基因组分析,R语言编程,Perl语言编程
往期精彩推文

