
Alphafold3:结构信息学的突破性技术革新

写在前面的
AlphaFold2问世后一度成为了领跑世界的蛋白质结构从头预测的标杆软件,Alphafold2在很大程度上极大降低了蛋白质建模和设计的成本,使得高通量大规模设计人工建模蛋白成为了可能。伴随Alphafold2的问世,其开发者DeepMind团队并没有停下继续革新软件的脚步,2024年5月8日,DeepMind团队在Nature发表研究性论文宣布了Alphafold2的迭代革新:AlphaFold3 模型,该模型在之前的基础上大幅更新了基于扩散的架构,能够联合预测包括蛋白质、核酸、小分子、离子和修饰残基在内的复合物的结构。与之前的所有软件相比(包括Alphafold2),Alphafold3在1.在蛋白质-配体相互作用中的预测准确性显著超过了现有的对接工具,2.在蛋白质-核酸相互作用上的预测准确性显著超过了其他工具,同时,Alphafold3在抗体-抗原预测准确性上显著超过了 AlphaFold-Multimer v2。该团队认为,在一个统一的深度学习范式框架内实现跨生物分子空间的高精度建模是可能的。
Alphafold2的劣势
虽然我的研究方向很少涉及到生物大分子结构预测,但是以前的部分研究还是些许涉及到了结构信息学相关分析。比如,预测蛋白和核酸的结合能力、氨基酸点突变对蛋白质局部空间结构的影响。由于Alphafold2底层算法及训练数据的有限性,使得以前的Alphafold2在我所知的如下方面具有较平庸的表现力。
1.AlphaFold2的局限性之一是它无法预测在真实世界中两个可以相互作用的蛋白的相互作用能力,已知的一些能相互作用的蛋白可以在相互作用的过程中产生柔性变化,进而使得这些蛋白在互作和不互作的两个状态下具有一些全局结构差异性。但是Alphafold2在这方面预测能力较差,也就是它只能较好地预测单独存在时蛋白的结构,这可能会影响蛋白质在互作状态下的的三维结构预测。
2.由于第一点提到的瓶颈,因此EBI的AlphaFold2数据库包含2亿个可以免费下载的结构预测,只包括了自然发生的蛋白质和其中的一个子集。它不包括自定义融合蛋白、合成蛋白、天然存在蛋白的突变、交替剪接版本或同种异构体。
3.对高性能Linux计算机的要求:AlphaFold2运行在本地机器上,需要安装在高性能、高容量的Linux计算机上,特别是对GPU计算能力的要求。这对于大多数无法获得此类资源的蛋白质研究人员来说是不切实际的。
4.复杂的设置和命令行用法:使用开源版本的AlphaFold2需要IT团队的帮助来进行设置,并且需要在命令行中输入复杂的命令。这对于不熟悉命令行脚本的用户来说绝对是一个不小的挑战。
缺乏蛋白质结构查看器:AlphaFold2的开源版本不包括蛋白质结构查看器。用户需要安装和学习额外的软件才能查看预测的蛋白质模型。
5.分子对接模拟的局限性:在分子对接模拟中,AlphaFold2可能无法准确地模拟蛋白质和药物之间的相互作用。所以在Alphafold2时代还有大量的结构生物学研究人员仅仅通过Alphafold2工具预测蛋白质结构,然后利用Rosetta或者pymol等软件导入蛋白质结构文件和小分子化合物文件进行二次预测蛋白和小分子化合物的对接能力。
6.较弱的氨基酸点突变预测能力:Alphafold2的优势在于更准确地进行全局蛋白质结构从头预测,但是对于因为氨基酸残基点突变发生的局部残基构象变化的分析,Alphafold2的能力稍逊于Rosetta软件。但是Rosetta软件的安装和命令行操作更复杂。
7.较弱的分子间结合自由能计算能力:Alphafold2在设计之初可能并没有考虑到做这方面的计算。但是结构生物学家常常有研究已知能够结合的生物大分子在其中一个有突变的情况下分子和分子之间的结合自由能是否会产生变化,如果有变化,这种变化是如何表征在结构的改变上。但是Alphafold2暂时没有这个功能,这使得当时结构生物学研究人员需要借助Amber等工具进行结合自由能相关的计算。
综上所述,Alphafold2的问世存在诸多短板,它主要是降低了大规模高准确性预测单个蛋白的全局结构的成本。如果要系统地解决生物大分子及其结构动力学和互作动力学的需求,需要Alphafold2做出较大更新。事实上,DeepMind团队早就考虑到了这些。
Accurate structure prediction of biomolecular interactions with AlphaFold 3
Alphafold3算法亮点
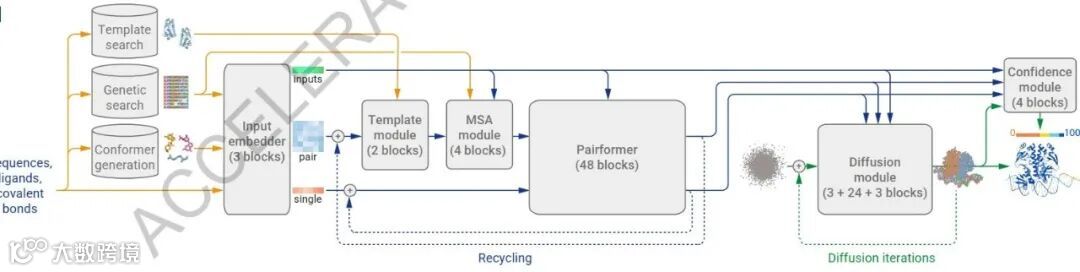
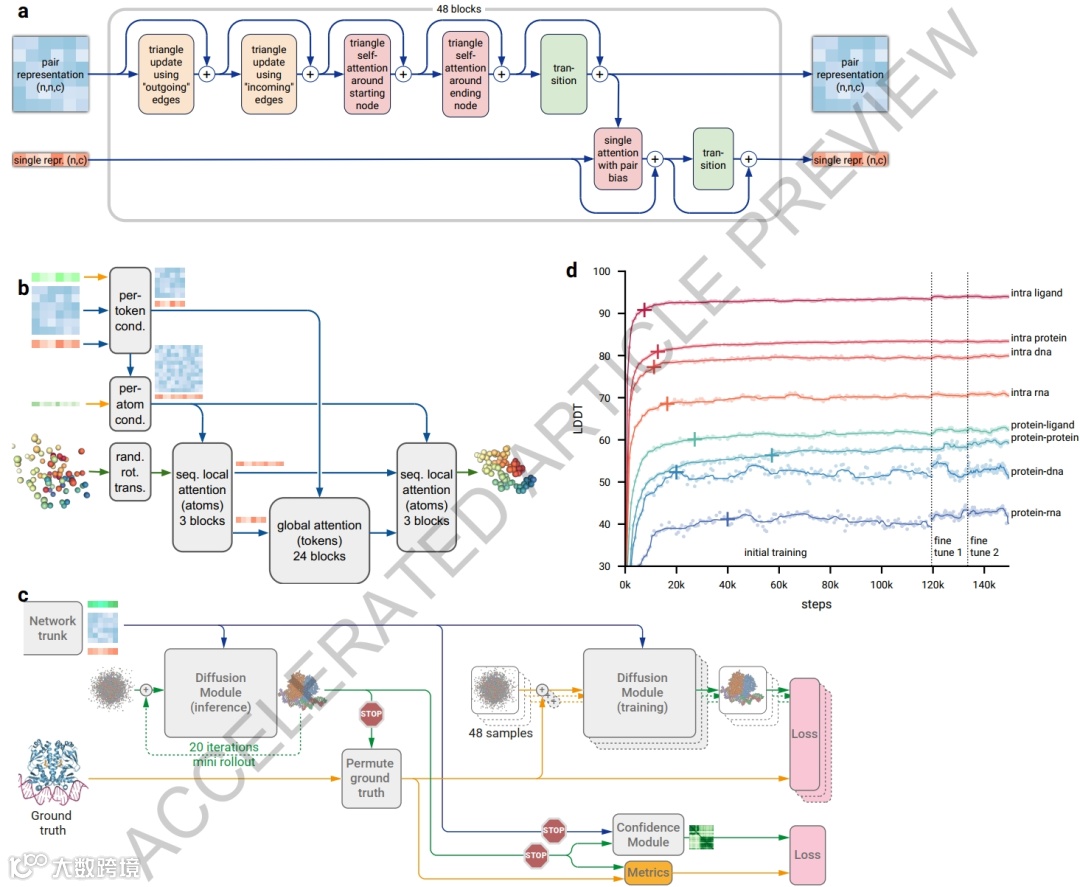
Alphafold2在蛋白质结构从头预测算法上除了使用了深度学习,同时还使用了一种叫共进化的算法,使得学习模型在预测结果时可以根据学习的过程而产生渐变式改进从而能尽可能地提高预测准确度。相比之下,Alphafold3的一大亮点是革新的扩散技术。这个技术的工作原理是通过训练一个模型,该模型从噪声图像出发,逐步减少噪声直到得到准确的预测。这种方法使得AlphaFold3能够处理更大规模的输入数据集。可以理解成AlphaFold3的能力源于其全新一代的架构和训练方式,其训练数据涵盖了所有生物大分子。该模型的核心是深度学习架构中改进版的Evoformer模块,能够为该模型的性能提供支持。在处理输入内容后,AlphaFold3使用扩散网络(diffusion)来解析每一个残基序列的原子,从单个原子出发,逐渐扩散,经过多个步骤,逐渐从原子到残基,残基到框架、框架到轮廓、轮廓到结构。最终以人工智能图像生成工具来处理整个预测过程,最终生成期望下准确度最大的分子结构。也正是因为扩散模型,使得生物大分子结构从头预测的准确度显著提高。
Alphafold3预测性能亮点
复杂复合体的预测性能
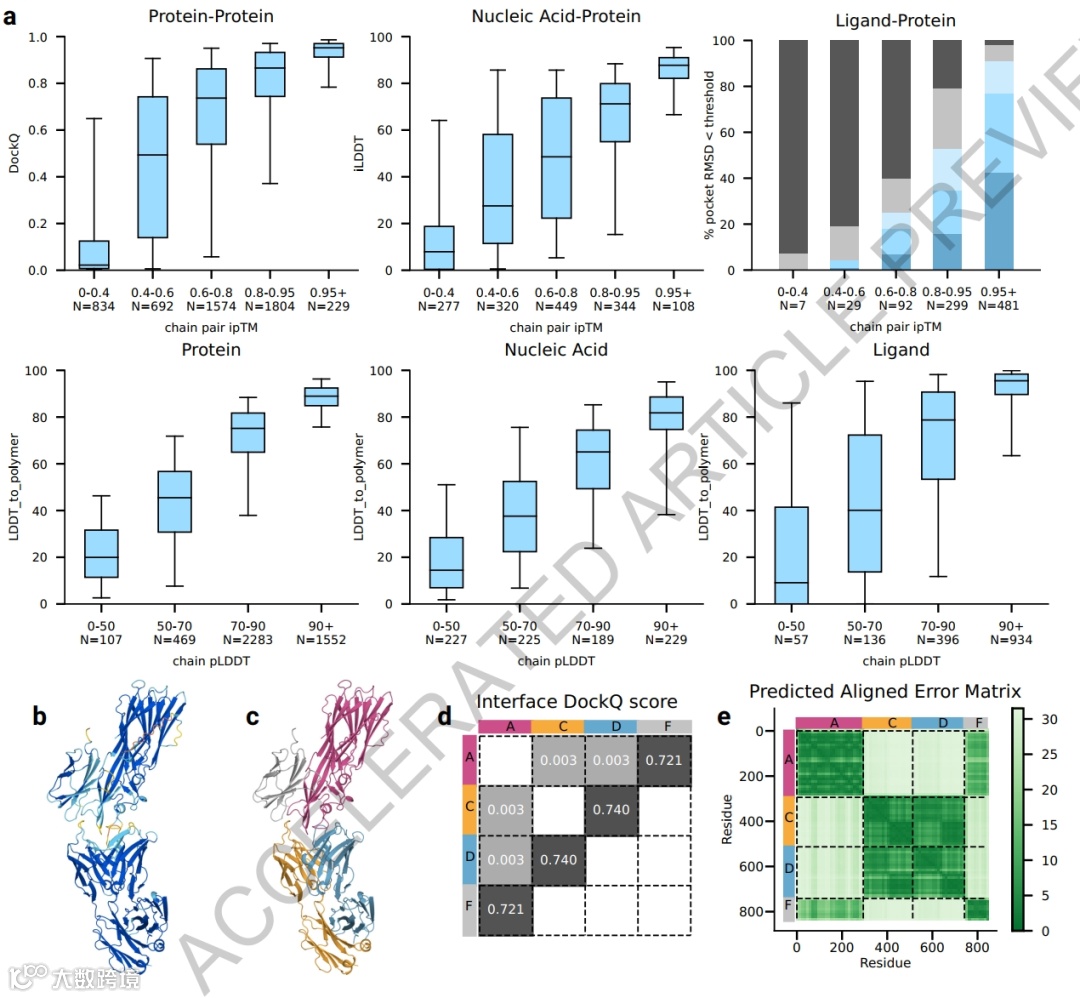

DeepMind团队利用已有高质量实验结构的PDB数据测试了Alphafold3对蛋白-蛋白,核酸-蛋白,蛋白-配体等复合体的预测,根据置信度作为评估策略,与以前的软件相比,其预测精度提高了一倍。根据该团队揭示的数据,AlphaFold 3 在预测各类分子的相互作用上体现出其他预测软件无法达到的准确性。并且这种预测模型只需要输入简单的一级序列信息或化学结构式。在某种意义上Alphafold3算得上第一个基于物理模型的生物大分子结构预测工具。除了对生物大分子的结构预测,Alphafold3还可以预测大分子基团上的共价修饰,这是Alphafold2无法做到的。利用高质量的PDB数据,以预测结构和真实结构的RMSD值为衡量指标,大部分经过Alphafold3预测的修饰与真实结构的RMSD小于2 Å,且ranking_model_fit > 0.5。DeepMind团队指出,Alphafold3在Alphafold2的基础上选择更轻量的MSA比对,尽管这一举措可以提高各类生物大分子的互作预测准确率,但是对于浅层MSA的蛋白质其预测准确度稍低。所以从这一方面,Alphafold3应该理解为Alphafold2在生物大分子及其相互作用动力学结构预测上的拓展,Alphafold3不能完全替代Alphafold2对蛋白质结构的预测。
Alphafold3的局限性
任何算法与模型的核心是通过数学和物理学的方式解释与逼近真实世界,由于Alphafold3本质依然是深度学习,它必然存在深度学习算法的一些瓶颈。所以使用者千万不能神化Alphafold3,就像Alphafold2的出现并没有让冷冻电镜失业一样。DeepMind团队在论文中已经提到了Alphafold3存在一些局限性,这些局限性涉及到立体化学(stereochemistry)、幻觉(hallucinations)、动力学(dynamics)及一些目标(certain targets)。
1.在立体化学方向,开发者已经注意到Alphafold3存在两类违规行为。首先,模型的一些输出结果并没有遵循化学中的手性原则。在立体化学领域,手性是指实物与其镜像不能在空间上重合的性质,人的左右手就是很好的例子,故而得名手性。由于手性分子广泛在自然界中存在,而Alphafold3的一部分输出结果(大约有4.4%的比例)不遵循手性原则,这使得结构生物学家试图利用Alphafold3研究旋光异构或非对映异构体时可能得到不真实的结果。为了消减Alphafold3输出结果的不可靠性,开发者团队在构建底层模型时试图引入手性罚分函数。尽管手性罚分函数可以在一定程度上降低不可靠结果出现的可能性,但是并不能做到根除这种结果的产生。另一方面,开发者也注意到Alphafold3在输出结果时偶尔会产生原子“碰撞”,顾名思义就是在空间中不同残基的原子在坐落在同一空间坐标中。开发者同样引入了原子冲突罚分函数,而这个函数依然只能降低,但是不能根除冲突现象产生。开发者指出在他们的测试数据中有大约100个核苷酸和2000多个蛋白质-核苷酸复合物都存在原子碰撞的预测现象。
2.虽然Alphafold3采用了扩散技术模型来提高准确预测生物大分子结构的种类,但是也正因为扩散模型,Alphafold3出现了结构幻觉现象。开发者团队注意到Alphafold3会在一些生物大分子的无规则区域引入虚假的结构基序。针对虚假结构基序的出现,开发者团队试图构建多模型预测排序模型,因为由幻觉产生的虚假结构通常具有更低的置信度,预测模型对同一个生物大分子预测后会产生多个可能的候选模型,候选模型会按照置信度进行排序。但是这种排序只能做到从概率的角度降低幻觉出现的几率,但是无法做到根除幻觉。
3.Alphafold3仍然没有解决Alphafold2无法解决的问题。就是对蛋白质在不同溶液环境中的结构预测。不同的生物大分子在不同的溶液体系中存在不同的系统动态行为。Alphafold系列的平台预测的结构只是表征蛋白质在静态环境中的结构。但是在溶液环境下,不同溶剂可能导致溶液的PH变化,而不同的PH对不同等电点的蛋白质有不同的电荷影响。所以在溶液环境中,蛋白质的结构属于非静态的。这一问题Alphafold2没有解决,Alphafold3也没有解决。
4.Alphafold3在进行结构预测时依然存在一些错误的情况。例如开发者已经发现,E3泛素连接酶在apo状态下通常是以开放构象存在,仅仅在靠近配体与其结合时会转变为闭合构象。但是Alphafold3却可以将无论是在holo状态下还是在apo状态下的E3泛素连接酶预测成开放构象。这已经与现有真实世界的结果相悖。开发者已经尽量在多序列比对后重采样过程中(这是Alphafold深度学习预测结构的一个前处理过程)做出了优化来降低这种情况的出现。不过这种瓶颈依然是存在的。
5.虽然Alphafold3已经极大拓宽了对不同类型的生物大分子预测的准确性。但是针对多目标(multi-targets)模型,特别是复杂的复合体结构,例如抗原抗体复合体,复杂蛋白复合物的预测存在精度的挑战。DeepMind团队建议增加预测输出的数量,例如产生1000个输出再进行排序比较以提高精度。但是这种做法非常耗费计算资源。一般实验室基本负担不起。所以要想用Alphafold3做出更好的结构预测。手里没有上百万的服务器硬件应该是比较困难的。
关于如何使用Alphafold3
目前DeepMind团队已经在新闻发布会中提及暂时没有公开完整源代码。他们提供了Alphafold3 Sever API。不过Github上有第三方团队根据Alphafold3公开的算法重构了Alphafold3模型,源代码可见Github(这还得感谢我的同事和我一起看这篇论文时他找的地址)。其实我很想搞清楚这个第三方团队的背景,这个代码是Alphafold3论文公开时一起公开的,目前代码刚刚公开不到24小时。
https://github.com/kyegomez/AlphaFold3
写在后面的
我相信当下会有许多推送会夸大鼓吹Alphafold3的性能。我在这篇推送中比较客观地解读Alphafold3的优势,也理性地指出Alphafold3存在的瓶颈。这些瓶颈并非我个人随便言语,我只是客观将他们发表的论文Limitation部分根据我的理解用中文转述了一次。在他们发表的Nature论文中,DeepMind团队花了很大篇幅详细地指出了Alphafold3的劣势和局限。我相信DeepMind团队在论文中如此详细地客观描写自己软件的局限也是想提醒学术界的研究人员不要过分相信Alphafold3的预测结果。诚然,Alphafold3已经在Alphafold2的基础上极大拓展了多种生物大分子预测的可行性和准确性,但是它仍然不是完美的。通过阅读他们的文章,我意识到DeepMind团队有意图打造Alphafold生态。他们今后的目的应该是想构建生物大分子的结构、动力学、互作等多维度深度学习范式生态。他们的目的不只是局限于结构的预测,他们的长远目的是将生物大分子(核酸、蛋白、糖、脂等化合物)多维度相互作用构建深度学习生态。用算法丈量生物大分子,抽象真实世界为数据世界,通过数据挖掘解读生命的底层架构。当然,现阶段仍然是非常初步的阶段。曾经许多人觉得Alphafold2的问世让做结构生物学的人都失业了,我想说的是,NGS和TGS的出现也并没有让做PCR的失业。相反PCR还因为高通量测序得到了巨大推动。Alphafold3的局限性同样也强调了传统基于实验的结构生物学测量方法依然占用重要的地位。当然,在这篇推送中我仅仅只是站在生物信息学的视角去理解Alphafold3对结构生物学的影响。这之中与结构生物学相关的知识可能有理解不到位的地方,如有错误,还望读者专家们批评指正。Alphafold3又是DeepMind团队领跑世界的一项大作,这篇论文相信会被很多专家和公众号解读。可以预计未来半年内会有大量关于测试Alphafold3在各种特殊复合体或生物大分子预测应用中的SCI论文发表,也会有大量手里有高性能服务器的公司或团队公开Alphafold3 Sever的付费服务。Alphafold3又会带动一批学术论文和盈利业务的产生。更相信的信息可以点击跳转阅读原文。