
如果我们在NCBI批量下载fasta数据,一般都是整合在一起的序列,这样会对我们的下游分析,比如基因预测、注释等带来麻烦。一条一条的单独下载fasta序列又会加大工作量,因此掌握在Linux下将整合的fasta序列进行拆分是很有必要的。
本文以一个例子来解释我的序列拆分过程,如下图:
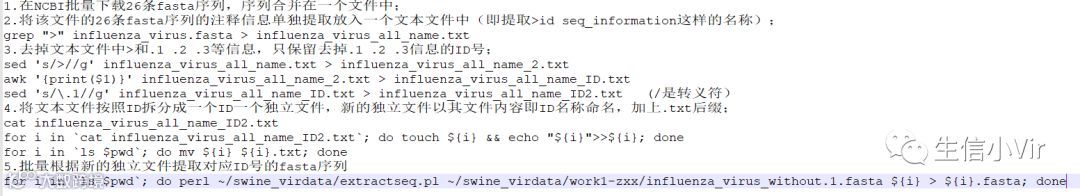
思路:1、grep提取fasta序列的信息;2、用sed把> 以及.1信息删除,在删除.1时,由于.在正则表达式中属于通配符,会把ID里所有的1同时匹配去除,因此使用sed删除.1时需要加上转义符\,将.的含义取消;3、只提取ID号,因为ID号和序列information之间有空格,因此可将其看作列表格式,用awk命令提取第一列即序列的ID号即可。4、把文本文件根据ID号进行拆分和命名,同时新的文本文件里的内容也是ID号。即用cat命令将influenza_virus_all_name_ID2.txt里的ID号输出到终端,用for循环批量创建文件(touch)并输入文件内容(echo);再把文件加上.txt后缀(mv)。5、最后用一个perl脚本提取对应ID号的fasta序列,注意需用引导符>指定输出文件。