
定向感知cfDNA
写在前面的
上一篇推文中提到,自从1948年科学家Mandel和Metias首次发现cfDNA后,在过去的70年中cfDNA相关领域被科学家逐渐探索并丰富。目前cfDNA是人类肿瘤早筛或产前诊断的重要生物标志物之一,基于cfDNA的筛查技术具有成为引领未来无创疾病诊断技术(NIPT)的巨大潜力。事实上,无论是疾病早筛还是产前诊断,发现标志性cfDNA的有无只是第一步。早筛的核心在于早发现、早治疗、早根除。如果能够从受检者采集的cfDNA样本中溯源出释放目标cfDNA片段的器官或组织,那么才能真正实现在疾病早期锁定病灶部位。特别是在肿瘤早筛领域,早期精准锁定病灶组织或器官,对于避免肿瘤转移、彻底根除疾病发生具有十分重大的意义。因此关于如何溯源cfDNA一直以来是该领域的重点和热点问题。
基于cfDNA末端信号的感知方法
今天要分享一篇来自卢煜明院士团队在2019年发表于《Genome Research》期刊的研究,这研究基于此团队过去在cfDNA领域的深耕结果,提出了一种在染色质开放区(Open Chromatin Regions, OCR)根据cfDNA双末端信号差异来推断cfDNA的器官来源的方法。在介绍这种方法之前,有三大基础知识点需要提前掌握:
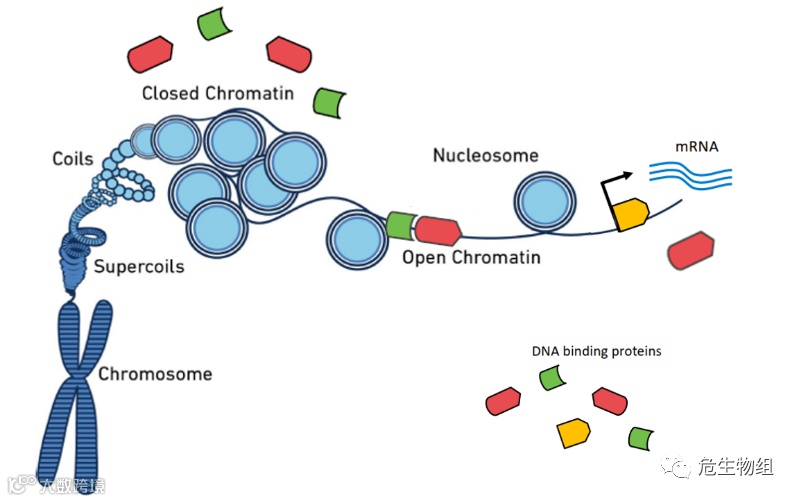
1.细胞核中人类DNA总长度约2米,如此庞大的DNA需要经过高度压缩才能储存于细胞核中,而DNA的压缩策略则是通过将DNA链按照约170bp为单位“缠绕”在大量由组蛋白构成的核小体上,形成复杂的高度折叠结构,每一个核小体之间存在大约20bp的非折叠DNA链作为连接两个核小体的linker。细胞中的DNA需要复制转录,而高度复杂的折叠结构使得DNA复制转录困难,因此高度折叠的染色质结构在复制和转录时需要在一些有利于起始转录复制的区域暴露DNA序列,这段暴露的序列就叫染色质开放区。如下图所示。
2.疾病发生早期通常伴随着免疫细胞识别清除病变细胞,细胞在凋亡后会释放较多cfDNA分子。在该团队前期工作已经发现,细胞凋亡后释放的DNA分子中,位于核小体之外的片段通常容易被核酸内切酶切割降解,而位于核小体上的DNA片段由于核小体的保护其降解半周期很长。
3.该团队早期的工作中已经建立了基于重亚硫酸盐测序(Bisulfite Sequencing, BS-seq)的方法推断cfDNA的贡献组织或器官。并且这种方法已经被其他实验室团队成功应用于推断cfDNA组织来源器官的研究中。因此证明了根据cfDNA特征推断组织来源是可行的。但是BS-seq测序成本相比于普通NGS高,且BS-seq会引入大量C>T转换和G>A转换,使得原始测序数据的碱基分布产生偏倚。因此有必要开发普适度更高、成本更低的方法来推断贡献cfDNA的组织或器官。
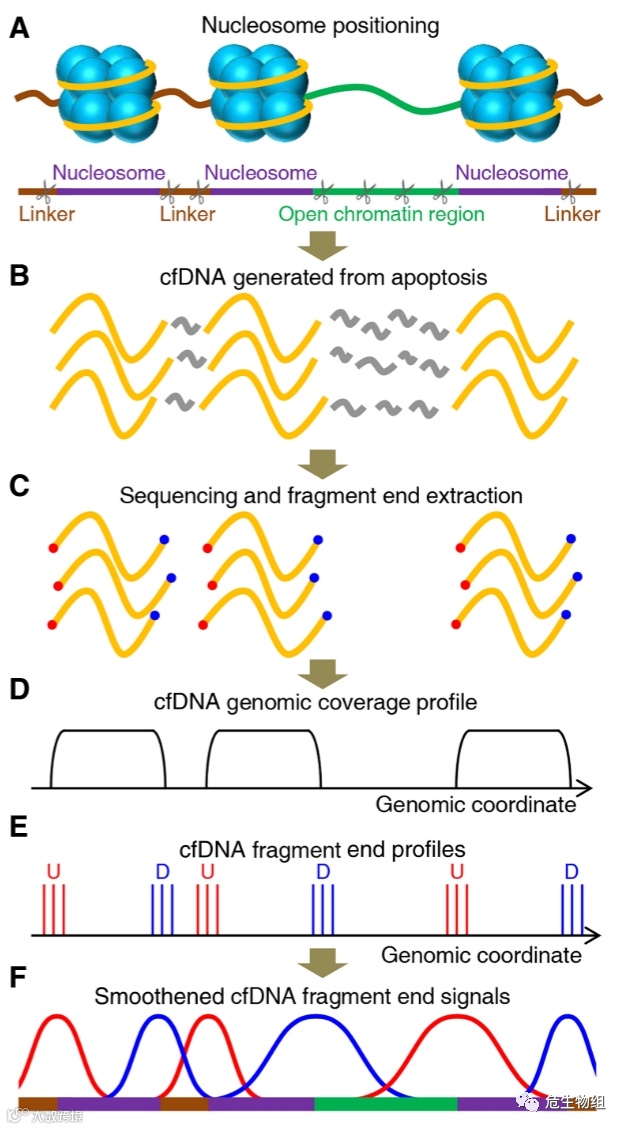
根据上面的基础知识,该团队做出第一个假设:如果细胞凋亡后释放的游离DNA分子中未被核小体保护的部分(linker或OCR)更容易被核酸内切酶切割进而降解,那么实际环境中linker或OCR对应的DNA片段的丰度就显著减少(下图B),而被核小体保护的DNA片段丰度则变化不显著,那么通过高通量测序后将cfDNA的测序reads映射回参考基因组后则会出现周期性测序深度高-低规律(下图D所示)。如果将任意一个核小体所对应的参考基因组区域视为一个模块,那么这个区域的5’端理论上会出现一个测序深度陡增的位点(U, Upstream end),3’端理论上也会出现一个测序深度陡降的位点(D, Downstream end)。那么理论上第i个D位点所对应的坐标值减去第i个U位点所对应的坐标值理论上应为第i个核小体DNA所在区间,第i+1个U位点所对应的坐标值减去第i个D位点所对应的坐标值理论上应为第i+1个核小体到第i个核小体之间的linker或OCR区域(下图E所示),如果前面的假设成立,那么每一个U end和每一个D end也是周期性出现,U end和D end也都能被拟合为周期信号曲线(下图F所示)。
为了验证第一个假设,该团队首先利用了自己在之前的科学研究中搜集的32名健康且非妊娠受试者的cfDNA测序数据。该团队首先选择了人类参考基因组12号染色体的一个区间,该区间被科学家报道为包含了几乎在人体所有类型组织中都出现的核小体。通过测序数据比对,果然证实了第一个假设,比对结果完美显示了周期性出现的190bp (170bp + 20bp)的信号规律。通过LOWESS拟合算法,这种周期性信号被完美展示。下图可以清晰地看出,不同周期中,相邻的D end与相邻的U end之间的距离差恰好约20bp,恰好对应linker的距离,同一周期中相邻的U end与D end之间的距离恰好约170bp,恰好对应核小体保护的DNA的长度。

基于组织特异性染色质开放区的cfDNA末端信号推断
当第一个假设被验证后,提示了基于cfDNA末端信号差推断cfDNA是从何种组织来源是可行的。但是仅仅基于第一个假设,还不能做到锁定cfDNA来源。由于染色质开放区域是基因复制转录前暴露的关键区域,而不同的组织器官受到不同的基因集调控,而不同的基因集坐落于基因组不同的位置,因此不同的组织器官一定对应了不同位置的特异染色质开放区(tissues-specific OCRs),因此该团队在这个基础上做出了第二个假设即在这些组织特异性的染色质开放区也存在着类似的cfDNA末端信号特征。根据前期的研究背景,该团队首先选择了T细胞和肝细胞共同的染色质开放区,这是因为一般情况下T细胞和肝细胞是临床上血液cfDNA的重要贡献细胞,因此该团队搜集了相关测序数据并研究了T细胞和肝细胞共有OCR区的cfDNA信号特征。如下图所示,将OCR区中心视为原点(图中0bp所在位置),OCR所在区域因为不含核小体(nucleosome-depleted),所以该区域U end和D end信号有急剧下降,但是在核小体区域,U end和D end就呈现了明显周期性信号(下图A)。从另一方面,该团队还选择了将测序数据映射至胚胎干细胞OCR,由于测序数据选自健康成年人队列,因此理论上胚胎肝细胞的OCR区域不存在类型信号特征,实际验证结果与理论预期符合(下图B)。通过这样的分析则提示出确实可以通过组织特异性cfDNA末端信号差异推断cfDNA可能的组织来源。
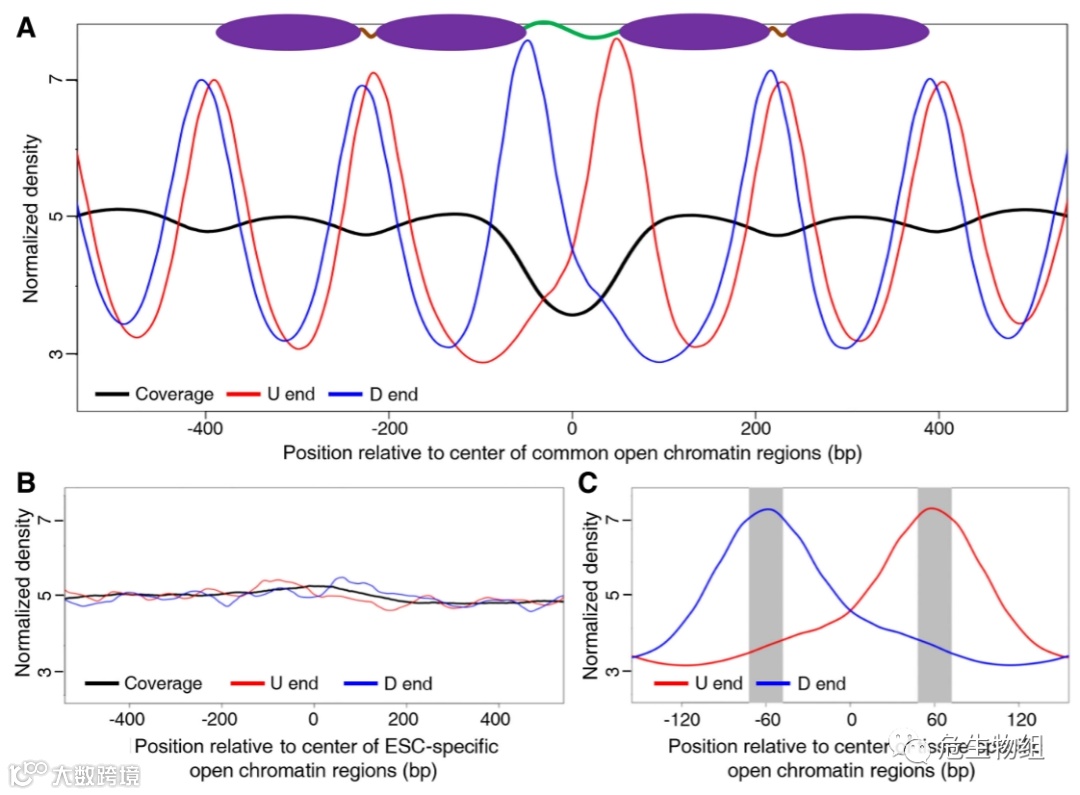
但是如何建立通用型判断方法呢?作者通过观察了他们测试数据的信号分布特征发现在染色质开放区中心的上游60bp,存在D end的峰值,也就是上一个核小体保护的cfDNA的3’端信号峰值,染色质开放区中心的下游60bp,存在U end峰值,也就是下一个核小体保护的cfDNA的5’端信号峰值。为了给予包容性,作者将这两个信号峰值各自的上下游10bp考虑进来,并根据下图3C建立了一个名为OCF计算公式的数学模型(Orientation-aware CfDNA Fragmentation)。看完这个公式,我明白这个数学模型的含义,本质上这个数学模型是在求解上图3C中阴影部分被U end和D end信号曲线围成的部分的面积(积分的思想),阴影部分面积越大(>>0)则代表OCF值越大,代表染色质开放区越呈现理想的cfDNA末端信号特征,也就说明cfDNA的来源组织越有可能是该组织特异性染色质开放区对应的那个组织,反之,如果OCF值接近于0或负数,则提示cfDNA来源于这些组织的可能性极小。其实看完这个公式后,我产生了一个疑惑,为什么是60bp,我猜测有可能是作者在论文中分析的这个数据恰好染色质开放区大约120bp,所以导致了上一个D end峰值和下一个U end峰值的坐标恰好位于其原点上下游60bp,所以对于任意染色质开放区,理论上公式中的60bp应该替换为染色质开放区的一半,或临近两个D end峰值对应坐标和U end峰值对应坐标的差值的一半。带着这个疑问我阅读了作者开发的预测程序的源代码,果然和我猜的一致,下面作者开发的预测程序的源代码中相对应的变量对应的默认值60,但是允许外部输入其他值。
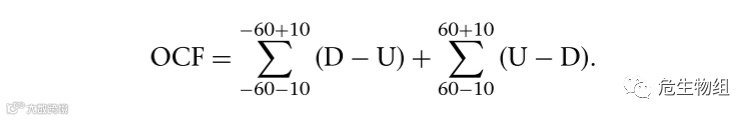
溯源模型的验证
根据前面的介绍,作者建立的数学模型应该会将一个测序数据映射至全基因组序列,然后逐一分析每个组织特异性染色质开放区的OCF值,最后统计汇总哪些OCF值显著大于0,则说明这些cfDNA可能来源于哪些组织器官。作者首先将他们的测试数据进行了分析,因为他们获取的是T细胞和干细胞的血液cfDNA样本的测序数据,最后结果也和实际吻合(下图)。
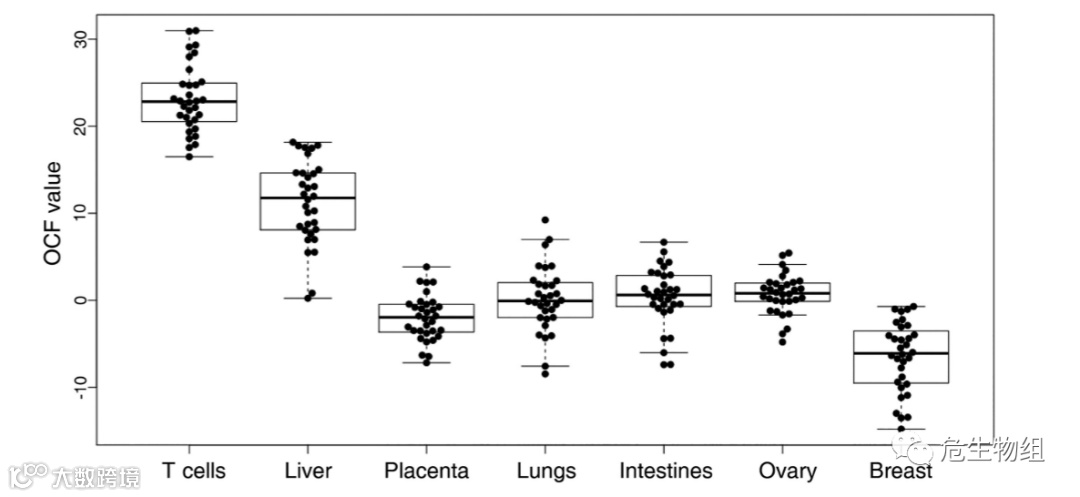
作者及团队进一步将构建的分析模型应用于无创产前诊断、肝移植、肝癌、结直肠癌和肺癌患者的测序数据中。均得到了有效验证和应用。其实在这个过程中作者并非仅仅进行了模型的应用,该团队在应用模型前通过公共大数据挖掘,分析出了人类全基因组多种组织器官组织特异性染色质开放区的信息,并构建了一个数据库。作者后续的应用均利用这个数据库进行分析推断cfDNA来源。
参考
Sun K, Jiang P, Cheng SH, Cheng THT, Wong J, Wong VWS, Ng SSM, Ma BBY, Leung TY, Chan SL, Mok TSK, Lai PBS, Chan HLY, Sun H, Chan KCA, Chiu RWK, Lo YMD. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res. 2019 Mar;29(3):418-427. doi: 10.1101/gr.242719.118. PMID: 30808726; PMCID: PMC6396422.
写在后面的
看完论文我感触颇深,一方面敬佩于作者所在团队长期在cfDNA领域的深耕,获得了非常深刻的科学积淀和认知,并在这个基础上巧妙地推理分析出更简单更高效的方法。一方面也敬佩该团队强大的交叉学科的技术背景,将全部的推测都进行了验证。但是在这篇论文的结尾作者提出了目前的方法只能定性研究,还无法做到定量研究。这让我想起来自己曾在博士期间研究的宏基因组检测未知病原技术,尝试了大量方法后发现所有的定量都只是尽可能地相对定量,完全绝对定量确实很难。