
在一项新的研究中,研究人员发现,一种新的小分子可以通过直接激活一种肿瘤抑制蛋白来阻止小鼠中的肺癌生长。相关研究近日结果在 Journal of Clinical Investigation 上,论文标题是“Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth”。
癌症的发展和进展涉及癌基因和肿瘤抑制功能的协调变化。特定的蛋白磷酸酶,如蛋白磷酸酶2A(PP2A),作为致癌信号的关键负调控因子,并受生理和病理机制的调控。 PP2A是一种丝氨酸/苏氨酸磷酸酶,可以控制许多细胞功能,如细胞周期,生长,代谢和凋亡。PP2A(protein phosphatase 2A)的磷酸酶能够通过移除附着到肿瘤蛋白上的磷酸分子来关闭这些蛋白的表达。根据研究表明这种肿瘤抑制蛋白的表达在几乎所有的主要癌症中都被关闭,其失活对于正常细胞转变成癌细胞至关重要。
基于此,该研究团队决定采用与以往不同的非常规方法进行癌症药物的开发,通过寻求直接靶向 PP2A 的分子,以便重新激活它的肿瘤抑制活性。研究人员通过对大量的化合物进行筛选,最后终于找到一组可以激活 PP2A的小分子化合物,并将其命名为 SMAPs,其原型药物为 reengineering tricyclicneuroleptics (图1)(Bioorg Med Chem. 2015, 23(19):6528–6534. Doi:10.1016/j.bmc.2015.07.007)。
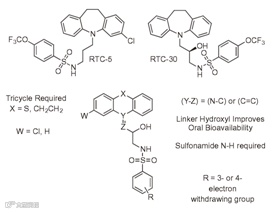
图1 经典的reengineering tricyclicneuroleptics结构
研究人员发现SMAPs 是通过附着到 PP2A 蛋白的一个亚基上,来有效地激活这种酶,这项研究也是首次利用一种小分子药物直接激活一种移除磷酸分子的酶。SMAPs 还防止肺癌细胞在实验室模型(包括临床前肺癌细胞模型和小鼠模型)中增殖。注射 SMAPs 的小鼠肺癌肿瘤会更少,而且也不会经历与其他癌症药物相关的体重减轻或行为异常。在小鼠模型中,SMAPs 与目前可用于减缓肺癌进展的组合疗法相当有效。
为了进一步确定 SMAPs 与 PP2A 结合的确切位置,这些研究人员培育出在假定的药物结合位点发生特定突变的肺癌细胞。由这些突变的肺癌细胞产生的肿瘤的小鼠如果并不受益于 SMAPs,那么这个位点即为 SMAPs 与 PP2A 结合的位点,因为在这种模型中 SMAPs 已经不能够结合和重新激活 PP2A。通过这种方式证实,SMAPs 在酶的亚单位内的两个特定氨基酸处与 PP2A 结合,这一发现将有助于其他药物开发。虽然这次的研究是以肺癌作为治疗的模型,但是该研究的科学家表示,由于 PP2A 失活是在很多癌症中普遍存在,因此除了肺癌,理论上这种 PP2A 的激活剂还可以用于治疗乳腺癌、前列腺癌、颅内恶性肿瘤、卵巢癌、子宫内膜癌等一系列的癌症。他们也正在准备下一步的临床试验,如果顺利的话未来人类将又掌握一个对抗癌症的利器。
请关注凌凯医药
