
大环化策略在药物设计中的应用与突破
提升药物亲和力、渗透性与稳定性,推动难成药靶点开发
本期文章共4709字,阅读时间约12分钟
大环骨架因其独特的理化性质,在药物设计中展现出显著优势。它不仅能增强化合物的生物活性和类药性,还可作用于传统小分子难以靶向的蛋白-蛋白相互作用等复杂靶标,突破Lipinski规则限制。近年来,随着结构生物学和合成方法的进步,经合理设计的大环药物数量持续上升,已成为新药研发的重要方向。
大环化合物介于传统小分子与大分子药物之间,具备独特的三维结构和构象刚性,赋予其更高的靶标亲和力、选择性、代谢稳定性及细胞渗透能力。基于靶标蛋白与配体共晶结构的理性设计,正成为大环药物开发的核心策略。
01 大环化显著提升药物亲和力
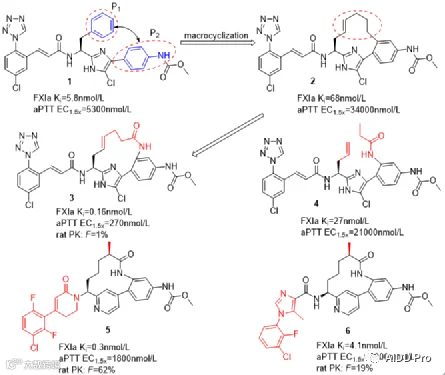
百时美施贵宝(BMS)在开发factor XIa抑制剂时,以苯基咪唑类化合物1为苗头分子。通过解析其与factor XIa的共晶结构,发现P1与P2结合域的空间邻近性,遂采用大环化策略,以脂肪醚链连接两区域,获得化合物2。进一步分析显示,烷基链靠近Leu41羰基,因此引入酰胺基团形成化合物3,成功建立氢键相互作用。晶体结构证实该氢键依赖于大环构象的稳定,使化合物3的Ki值达0.16 nmol·L⁻¹,活性较非环状类似物提升近160倍。尽管初始化合物口服生物利用度低(1%),后续优化已获得具备良好口服活性的候选分子5和6。
02 大环化改善药物渗透性
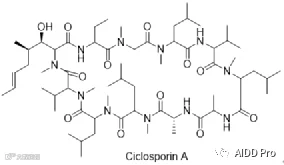
研究发现,20种可口服的大环药物普遍具有“变色龙效应”——能根据环境极性调节自身极性,降低极性表面积以适应疏水环境,从而增强跨膜能力。环孢菌素A是典型代表,其体外透膜速率仅为2.5×10⁻⁷ cm·s⁻¹,远低于常规药物标准(10⁻⁵ cm·s⁻¹),但仍可有效穿透细胞膜。核磁共振揭示其在水相与有机相中采用不同构象,通过分子内氢键网络实现构象切换,促进膜渗透。这一机制广泛存在于天然环肽类大环化合物中。
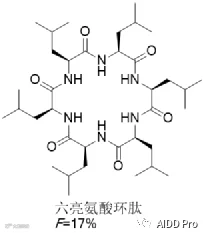
天然环肽为大环药物设计提供了丰富模板。通过引入杂环、酰胺甲基化或疏水屏蔽等手段构建分子内氢键,可减少与水分子的氢键作用,提升渗透性。六亮氨酸环肽即为一例:其分子量679、氢键供体6个、受体12个、cLogP为9.1,明显违背Lipinski规则,但由于疏水侧链屏蔽了极性酰胺基团,仍具备17%的口服生物利用度。
03 大环化增强药物稳定性
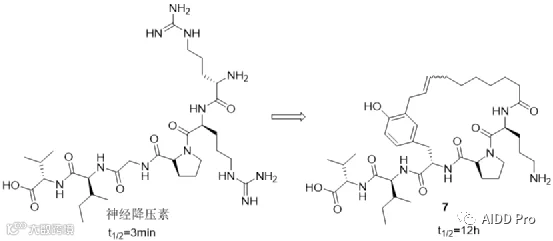
环肽因空间位阻和关键位点被包埋,可显著提升线性肽的代谢稳定性。神经降压素是一种具有强效镇痛活性的十三肽,其活性片段为C端六肽,但线性结构导致口服无效且体内半衰期不足5分钟。通过将第9位精氨酸替换为赖氨酸,并以烷基链替代不稳定的第8位残基进行大环化,获得化合物7,血浆半衰期从3分钟延长至12小时,稳定性大幅提升。
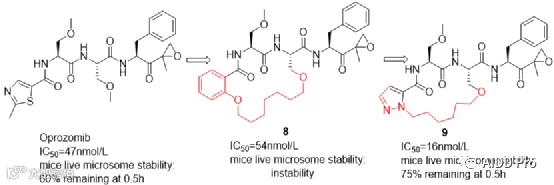
Oprozomib是一种处于I/II期临床试验的蛋白酶体抑制剂。结构研究表明其O-甲基丝氨酸与噻唑环空间接近,但前者无直接结合作用。据此进行大环化得到化合物8,虽保持原有结合模式,但稳定性未改善。进一步优化发现,成环末端采用含氮五元杂环更有利于构象稳定。以吡唑环替代苯环后获得化合物9,活性与代谢稳定性均显著提升。
04 大环合成的关键挑战与应对策略
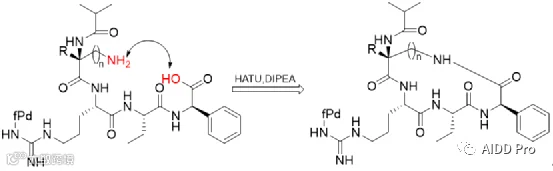
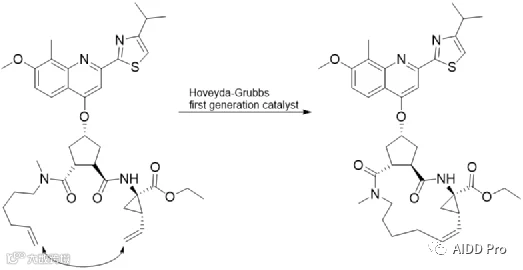
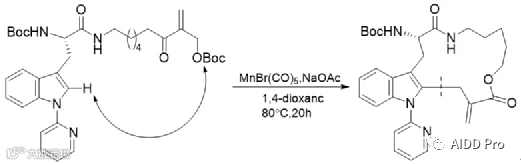
大环化策略应用受限的主要瓶颈在于合成难度高,尤其是关环反应常面临低产率问题。由于需克服分子内反应的熵损失,通常需高稀释条件以抑制分子间副反应。为此,合成化学家发展了多种高效关环方法,主要包括酰胺化或酯化、Click反应、烯烃复分解及C-H活化等。

05 小结
面对日益复杂的药物靶标,尤其是蛋白-蛋白相互作用等“难成药”靶点,传统小分子设计面临瓶颈。大环化合物凭借其独特的空间构型和理化特性,展现出在亲和力、选择性、代谢稳定性和细胞渗透性方面的综合优势。尽管早期受限于合成技术,大环结构多见于天然产物,但随着药物化学与合成方法的进步,理性设计的大环药物不断涌现,并已有多个成功上市案例,验证了该策略的可行性与前景。未来,随着合成技术的持续突破,大环化将在创新药物研发中发挥更加重要的作用。