通讯单位:北京大学,中国科学技术大学,上海交通大学
论文DOI:https://doi.org/10.1038/s41467-022-34463-7

全文速览
通过调节强金属-载体相互作用(SMSI)来调整界面催化剂的几何和电子结构已经引起了相当大的关注。本文报告了一系列 TiO2-x/Ni 催化剂。其中,无序的 TiO2-x覆盖层固定在 Ni 纳米颗粒(~20 nm)的表面上,并具有SMSI 效应。在 220 °C 大气压下的费托合成 (FTS) 过程中,最佳的TiO2−x/Ni 催化剂的 CO 转化率约为 19.8%。更重要的是,约 64.6% 的产物是 C2+ 链烷烃,这与主要产物是甲烷的传统 Ni 催化剂的结果形成鲜明对比。先进的电子显微镜、原位光谱表征和密度泛函理论计算研究表明,存在的 Niδ-/TiO2-x 界面位点,可以牢固地结合碳原子,抑制甲烷的形成并促进 C-C 链的生成,导致在 Ni 表面生成 C2+ 烃。

背景介绍
通过费托合成(FTS)工艺将CO加氢生成高附加值化学品受到广泛的关注,并已成为学术界和工业界最前沿的领域。尽管该过程在工业上取得了巨大成功,但是,Fe 或 Co 基 FTS 的反应机理和构效关系仍然存在很多争论。寻找 Co 或 Fe 催化剂的替代品,以及从新的角度对结构-活性关系进行阐释至关重要。长期以来,Ni 基催化剂一直被认为是 FTS 的重要候选者,因为与 Co 或Fe 催化 FTS 相比,它们在相对温和的工作条件下表现出较高的 CO 加氢活性。但是,甲烷化和由于形成Ni-羰基而导致的快速失活,严重阻碍了其实际应用。目前,一些实验和计算报告了金属-载体相互作用及其对镍催化 FTS 中产物分布的影响,这表明最佳的镍基催化剂可能表现出与钴催化剂相似的 FTS 活性。因此,开发一种新型的镍基催化剂以有效抑制羰基镍的形成并同时增强C-C链的增长,从而提高CO加氢对长链烃的选择性具有重要意义。据报道,调节 Ni 颗粒的几何和电子结构应该是调整 CO 加氢对长链烃的催化性能的有效方法。
研究人员提出了强金属-载体相互作用(SMSI)。它改变了负载金属颗粒的几何和电子结构,尤其是界面特性。低氧化物在负载金属颗粒表面迁移,在界面处具有强电子转移,导致原子重新排列或界面电荷重新分布,这是 SMSI 的典型特征。实际上,研究人员已经开发了许多 SMSI 型催化剂,以提高 CO 加氢中所需长链烃的催化活性和选择性。例如,由于位于 Ru/TiO2界面的 TiO2-x 覆盖层促进了 CO键的活化和断裂,因此在 FTS 期间通过 SMSI 也可以有效地提高 Ru/TiO2 催化剂的催化性能。与传统的浸渍方法相比,层状双氢氧化物 (LDHs) 的原位结构转变可以为构建新型 SMSI 催化剂提供一种有效的前沿方法,这在近年来的 C1 催化化学中得到了探索。这些结果提供了一些指导,用于合成一种新型的镍基 SMSI 催化剂,其具有增强的 FTS 性能。

图文解析

图1. 在不同条件下,TiO2−x/Ni 催化剂在合成气转化中的结构性质和催化性能。a NiTi-MMO 和一系列 TiO2-x/Ni 催化剂在不同温度下还原的 XRD 图谱。b 不同 TiO2-x/Ni催化剂的转化率和产物分布,以及 (c) 反应速率和活性。d TiO2−x/Ni-450 在 220 °C 下的稳定性评估。反应条件:催化剂(120 mg),1 bar,合成气(CO/H2/Ar = 32/64/4;空速:10000 mL gcat-1 h-1)。

图2. 不同 TiO2−x/Ni 催化剂的几何结构和电子结构表征。不同 TiO2-x/Ni 催化剂的 (a) Ni 2p 和 (b) Ti 2p 的准原位 XPS 光谱。c 在室温下,TiO2-x/Ni 催化剂的原位 CO-DRIFTS 光谱。d 不同TiO2-x/Ni 催化剂的归一化 Ni K-edge XANES 光谱。e 相应的归一化 Ni K-edge 傅里叶变换 EXAFS 光谱。f 不同 TiO2-x/Ni催化剂的标准化 Ti K-edge XANES 光谱。(g) Ni 箔、(h) NiTi-MMO 和 (i) TiO2-x/Ni-450催化剂的 WT 分析。
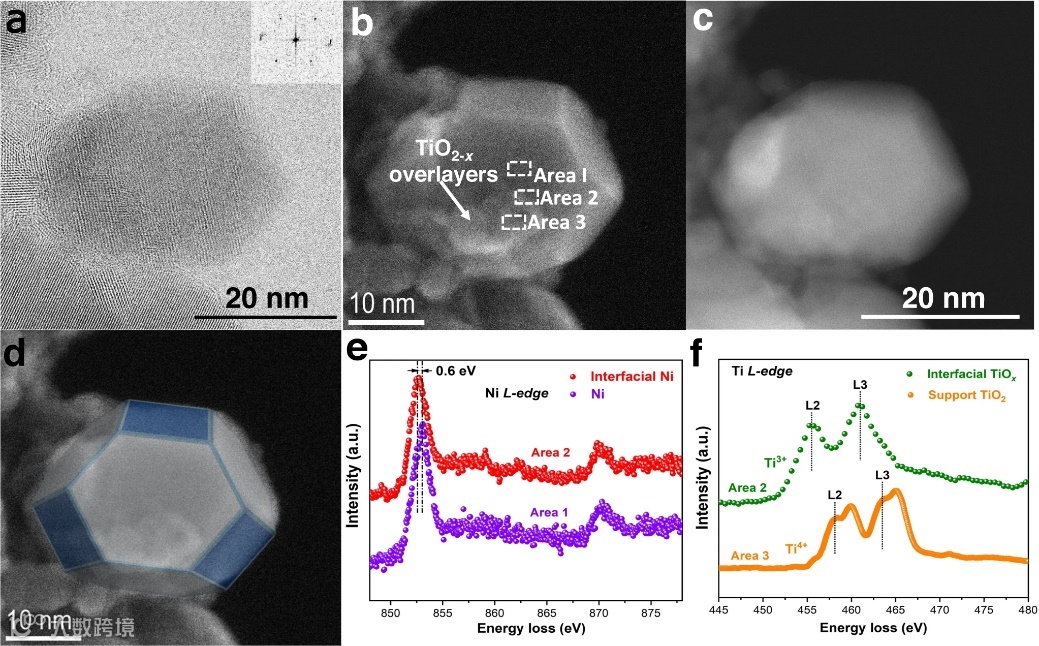
图3. TiO2−x/Ni-450催化剂的结构和形貌表征。TiO2−x/Ni-450催化剂在 H2 气氛 (10 Pa) 中450 °C 还原后的 (a) STEM-BF、(b) STEM-SE 和 (c) STEM-ADF 图像。d Ni颗粒示意图。在点 I 和 II 收集的原位电子能量损失光谱 (EELS),包括 (e) Ni L-edge 光谱和 (f) Ti L-edge 光谱。

图4. 通过原位红外光谱研究CO加氢机理。在 TiO2−x/Ni-450催化剂上,分别在 a、b 160 °C、c、d 180 °C、e、f 200 °C、g、h 220 °C 下获得的原位时间分辨 DRIFTS 光谱。
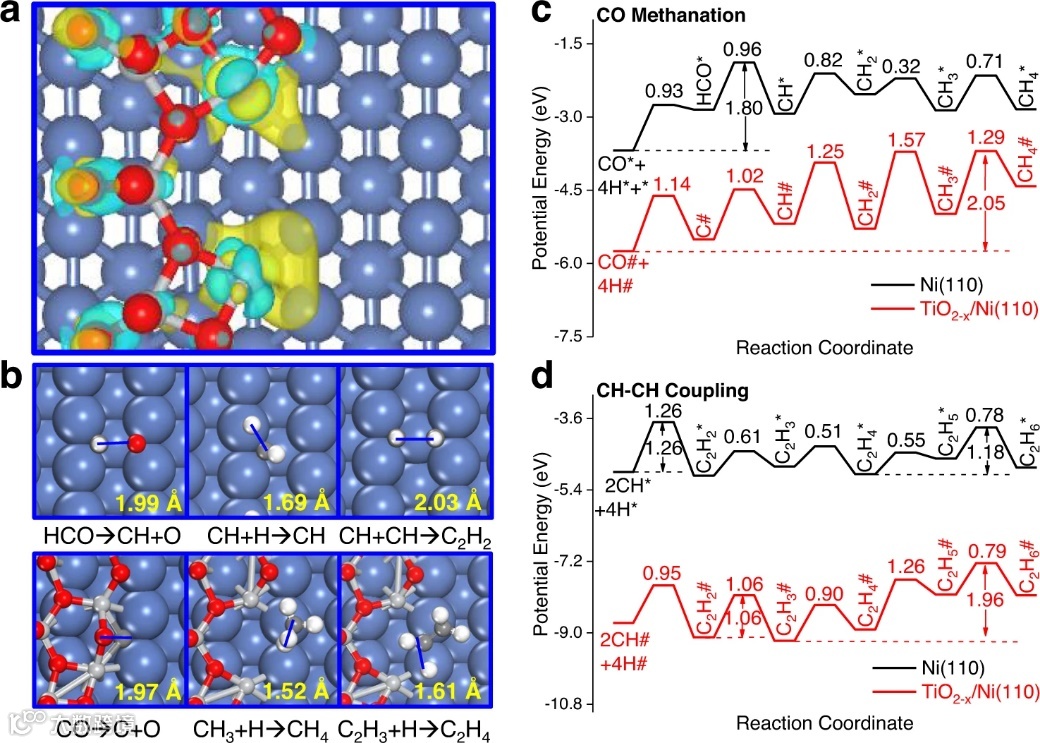
图5. DFT优化结构和反应机理研究。a Ti6O11/Ni(110) 表面的微分电荷密度。浅蓝色和黄色轮廓(等值面 = 0.006 e/Å3)分别代表电荷耗尽和电荷积累。b 在 Ni(110)(上图)和 Ti6O11/Ni(110)(下图)表面上 CO 加氢生成 CH4 和 C2H6 产物的关键过渡态结构。绘制的蓝色实线表示键解离或形成。CO氢化的过渡态的键距离以Å表示。蓝色、浅灰色、灰色、红色和白色球体分别是 Ni、Ti、C、O 和 H 原子。c, d 分别在 Ni(110) 和 Ti6O11/Ni(110)表面上,通过 CH-CH 偶联和氢化实现 CO 甲烷化和 C2H6 合成的势能图。

总结与展望
综上所述,本文基于LDHs 前驱体制成了一种 TiO2−x/Ni 催化剂,其在 FTS 工艺中表现出约 19.8% 的高 CO 转化率和约 64.6% 的 C2+选择性。电子显微镜和原位表征验证了无序二氧化钛覆盖层可以通过 SMSI 调节 Ni 颗粒的表面,以产生丰富的 Niδ-/TiO2-x界面位点。DFT计算表明,TiO2-x/Ni催化剂对CO加氢所表现出非凡的活性和选择性源于催化协同效应,即界面Niδ-/TiO2-x位点增强了CO解离,促进了C-C链的生长。本工作不仅发现了一种具有独特界面结构的新型SMSI催化剂,用于常压下合成气转化,还深入了解了SMSI效应驱动的界面协同催化。
本文仅供科研分享,不做盈利使用,如有侵权,请联系后台小编删除
欢迎关注我们,订阅更多最新消息 “邃瞳科学云”推出专业的自然科学直播服务啦!不仅直播团队专业,直播画面出色,而且传播渠道多,宣传效果佳。
“邃瞳科学云”推出专业的自然科学直播服务啦!不仅直播团队专业,直播画面出色,而且传播渠道多,宣传效果佳。
“邃瞳科学云"平台正在收集、整理各类学术会议信息,欢迎学会、期刊、会议组织方择优在邃瞳平台上进行线上直播,希望藉此帮助广大科研人员跨越时空的限制,实现自由、畅通地交流互动。欢迎老师同学们提供会议信息(会有礼品赠送),学会、期刊、会议组织方商谈合作,均请联系翟女士:18612651915(微信同)。
投稿、荐稿、爆料:Editor@scisight.cn







