


第一作者:胡海均
通讯作者:孙晓东
通讯单位:辽宁大学清洁能源化学研究所
论文DOI:10.1002/aenm.202303638

共价有机骨架(COFs)具有共轭度高、结晶度好、孔隙率大、结构可调控等优点,近年来已成为光催化制氢领域备受关注的材料。然而COFs较低的电子-空穴分离效率和缓慢的电荷迁移速率使其难以达到较高的光催化活性。异质结的构建被认为是一种有效的策略,但异质结的形成通常会受到杂化材料维度不匹配的影响,甚至以牺牲材料的氧化还原能力为代价。为了解决这些问题,利用H2N-Cu-MOF(NCM)和TpPa-1-COF(TP1C)构建一种新型的2D/2D S型异质结可以实现高效光解水制氢。两种晶体多孔材料的维度匹配使得NCM/TP1C杂化材料具有丰富的表面反应位点,面面接触使得两种材料发生强相互作用,并且紧密的层间电子相互作用优化了电子结构。此外,S型异质结的构建不仅可以促进载流子的分离,还可以使得材料具备较高的氧化还原能力,从而显著提高其光催化活性。结果,20% NCM/TP1C在不添加助催化剂条件下的光解水产氢速率可以达到4.19 mmol·g-1·h-1,远远超过了NCM和TP1C。

随着经济的发展和人口的增长,能源短缺和环境污染问题日趋严重。氢能是新一代具有远大前景的可持续清洁能源,被认为是太阳能储存的理想载体。因此,设计高活性、高稳定的光催化剂对氢气的制备至关重要。共价有机框架材料(COFs)具有低密度、高结晶度、孔结构可调控、易于改性等独特优点。然而,由于材料中电子-空穴对的严重复合,纯COFs的光催化性能相对较差。金属有机框架材料(MOFs)是一种具有拓扑结构的多孔材料,它是由金属离子和有机连接配体组装而成。与传统的无机材料相比,MOFs具有高比表面积、超高孔隙率、活性中心丰富、结构可调等特点,而带隙宽、光吸收范围窄等缺点限制了其在光催化领域的应用。一般来讲,异质结的构建可以有效提高催化剂的活性,而异质结的构建经常遭遇维度不匹配的难题,且传统异质结的构建降低了催化剂的氧化还原性,解决这些难题是制备高活性催化剂的关键。

3. 原位ESR测试、原位XPS测试和DFT计算的结果证明了S方案异质结的成功构建,这不仅保证了NCM/TP1C杂化材料的高氧化还原能力,而且进一步促进了光生载流子的分离。因此,由于独特的2D/2D晶体多孔结构和S型电荷转移途径,所构建的NCM/TP1C异质结表现出高的光催化性能。

本文首先通过溶剂热法分别制备了NCM和TP1C材料,然后通过研磨和超声法制备了NCM/TP1C杂化材料(图1a)。从SEM图中可以看出,NCM和TP1C纳米片分别堆叠而呈现出花状和层状形貌(图1b-c)。通过简单的研磨和超声处理,两种晶体材料通过静电相互作用成功地结合在一起(图1d)。此外,SEM-EDS测试进一步证明了NCM/TP1C杂化材料的成功制备(图1g-j)。
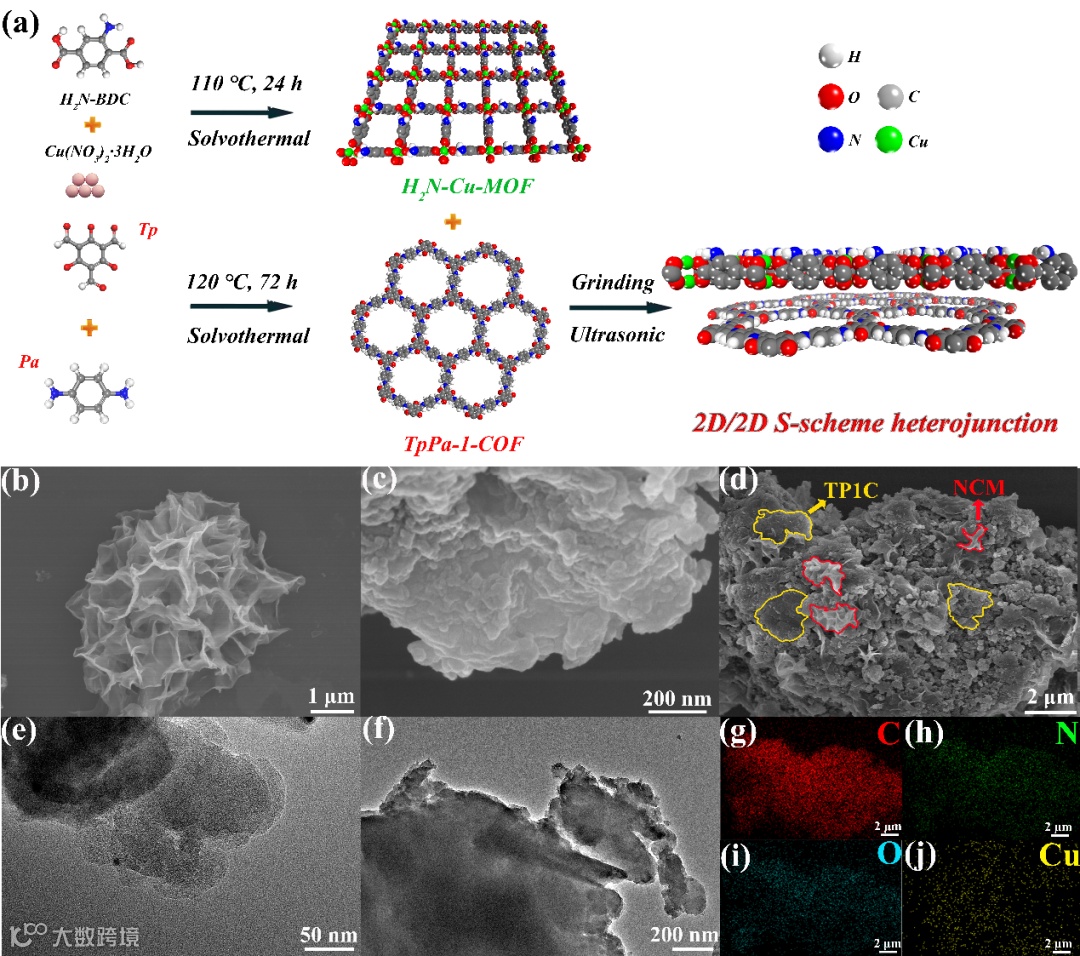
图1 (a)NCM/TP1C杂化材料的合成示意图;(b-d)NCM, TP1C 和20% NCM/TP1C的SEM图;(e-f)TP1C 和20% NCM/TP1C的TEM图;(g-j)20% NCM/TP1C的SEM-EDS图。
通过X射线光电子能谱测试(XPS)表明NCM材料中Cu2+的存在,并证明了NCM/TP1C杂化材料的成功制备(图2a-b)。紫外-可见漫反射光谱(图2c)表明,当NCM与TP1C结合后,材料的光吸收范围变大。结合Kubelka-Munk方程及莫特-肖基测试等表征对NCM和TP1C的能带结构进行分析(图2d-f),结果表明,NCM与TP1C的能带结构错位排列,两种材料合适的能带位置使得S型异质结的构建成为可能。
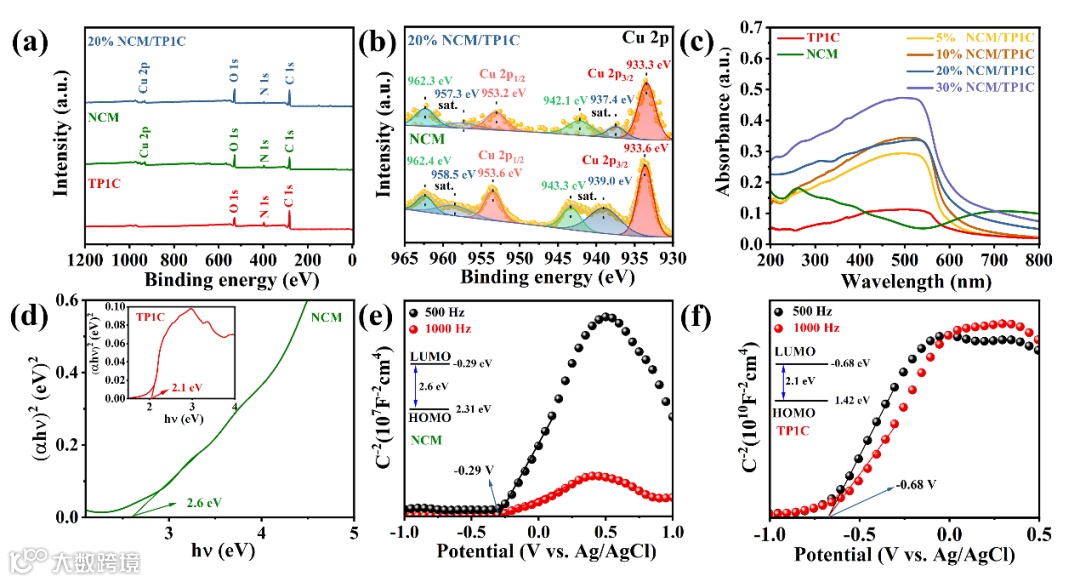
图2 (a-b)所制备样品的XPS光谱图;(c)所制备样品的紫外-可见漫反射光谱图;(d)NCM和TP1C的Tauc图;(e-f)NCM和TP1C的莫特-肖基图。
如图 3a,由于较窄的光吸收范围及较高的载流子重组率,NCM和TP1C都表现出较低的光催化产氢活性,而当两种材料结合后,光催化性能明显提升。当加入助催化剂Pt后,材料的催化性能进一步提高(图 3e),并超过了许多已报道的光催化剂(图 3g)。循环测试表明,NCM/TP1C材料具有良好的光催化稳定性(图 3d)。将杂化材料在不同牺牲剂中进行了光催化测试,结果表明,当抗坏血酸作为牺牲剂时NCM/TP1C材料的催化性能最佳(图3f)。

图3 (a-b)所制备催化剂的光解水产氢量及光解水产氢速率;(c)NCM/TP1C杂化材料的表观量子效率;(d)NCM/TP1C杂化材料的循环性能测试图;(e)助催化剂Pt对催化剂性能的影响;(f)不同牺牲剂对催化剂性能的影响;(g)NCM/TP1C杂化材料与已报道的光催化剂的性能对比。
从原位ESR测试图(图4a)可以看出,20% NCM/TP1C的信号强于TP1C,结合两种材料的能带结构的分析,表明NCM/TP1C异质结符合S型电荷转移机理。原位XPS测试表明,在光照条件下,电子从NCM流向TP1C(图 4b-c)。此外,DFT计算表明,与TP1C相比,NCM具有更大的功函数,在无光照的情况下,电子从TP1C流向NCM(图 4g-h)。电荷密度差分图进一步证明,在无光照条件下,电子从TP1C流向NCM(图4 f,i)。因此,结合原位ESR测试、原位XPS实验和DFT计算结果,证明NCM/TP1C材料符合S型光催化反应机理。

图4 (a)TP1C及20% NCM/TP1C的ESR测试图;(b-c)所制备材料的原位XPS测试;(d-e)NCM和TP1C的分子结构示意图;(f-i)所制备材料的DFT计算。
催化剂的反应机理如图5所示,NCM与TP1C结合后,由于功函数的差异,电子从TP1C流向NCM,直至费米能级达到平衡,这导致材料内部能带弯曲并产生内建电场。光照条件下,由于内建电场的作用,NCM的最低未占据分子轨道(LUMO)上的光生电子与TP1C的最高占据分子轨道(HOMO)上的光生空穴发生重组,TP1C的LUMO上的电子将水中的H+还原为氢气,而NCM的HOMO上的空穴则被牺牲剂消耗掉。
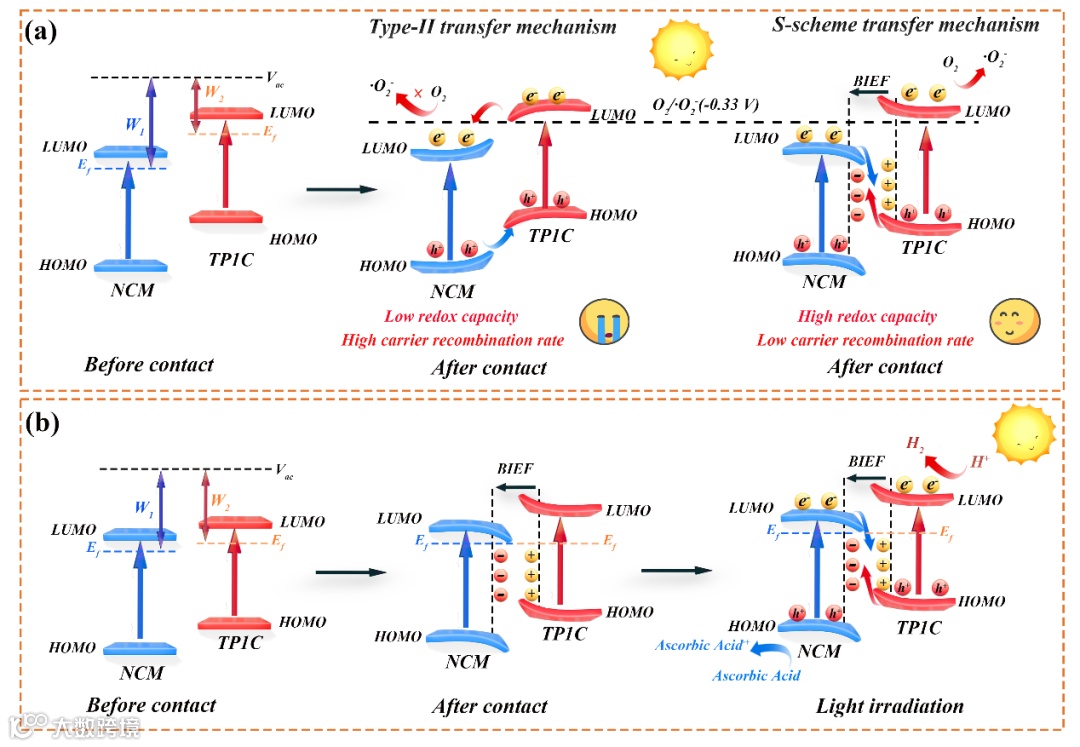
图5. NCM/TP1C杂化材料的光催化机理图。

本工作成功构建了基于晶体多孔材料的2D/2D S型异质结用于光催化水分解制氢。首先,通过溶剂热法分别制备了NCM和TP1C,然后通过研磨超声法制备了NCM/TP1C杂化材料。由于两种晶态多孔材料均为二维结构,能够很好地进行维度匹配,它们可以通过静电相互作用建立面对面的紧密接触,这可以提供更多的反应位点并有利于电荷转移。此外,通过原位测试和DFT计算证明了该杂化材料的S型电荷转移机制,这不仅可以缓解载流子的重组,还可以使杂化材料保持较高的氧化还原能力。因此,NCM/TP1C杂化材料的光催化析氢速率高达4.19 mmol·g-1·h-1,远远超过了NCM和TP1C材料的催化产氢速率,并且优于多数已报道的基于COFs的光催化剂。这项工作为构建基于二维晶态多孔材料的新型S型光催化体系提供了新的方法。

胡海均,辽宁大学博士研究生,主要研究方向为基于基于晶态多孔材料光催化剂的设计及光解水制氢性能的研究。目前已分别在Advanced Energy Materials,Chinese Journal of Catalysis和Dalton Transactions等期刊上以第一作者发表SCI论文3篇。
孙晓东,辽宁大学,清洁能源化学研究院,助理研究员,硕士生导师。主要研究方向为晶态多孔材料及其在能源催化领域的应用。以第一作者或通讯作者在Angew. Chem. Int. Ed.,Adv. Funct. Mater.,Adv. Energy Mater.,Nano Energy,Small等期刊上发表SCI论文20余篇。多次获得沈阳市及辽宁省自然科学学术成果奖,2023年获得英国皇家化学会“Horizon Prize”。Inorganic Chemistry Communication,Coordination Chemistry Reviews,Chemistry-An Asian Journal杂志特邀审稿人。获得国家自然科学基金项目1项,辽宁省自然科学基金项目1项。

Haijun Hu, Xinyu Zhang, Kailai Zhang, et. al.Construction of a 2D/2D Crystalline Porous Materials Based S-Scheme Heterojunction for Efficient Photocatalytic H2 Production. Adv. Energy Mater. 2024, 2303638.
https://doi.org/10.1002/aenm.202303638
声明

“邃瞳科学云”直播服务

扫描二维码下载
邃瞳科学云APP



