


当前,全球对能源安全、经济和环境的关注日益加剧,源于对石油的过度依赖。生物质转化通过裂解碳-氧(C-O)键,可提供石油基化学品和燃料,其中木质素是一种丰富的可再生资源,能用于生产高附加值的醚类化合物。然而,传统方法从化石资源中提取木质素存在能耗大和污染严重的问题。电催化技术通过改变催化剂表面化学势,促进目标化学转化,提供了一种高效解决方案。尽管已有研究在催化剂设计上取得进展,未来仍需探索高效的催化方法,以实现选择性裂解C-O(H)键并保留醚C-O(R)键。
在此,中国科学院化学研究所韩布兴院士课题组报告了一系列介孔碳支撑的铂-钴(Pt-Co)双金属电催化剂,其中铂和钴之间不同的金属间相互作用导致了不同的氢脱氧和氢化路线。值得注意的是,用 Co 单原子装饰的铂纳米粒子可选择性地裂解愈创木酚和其他木质素衍生物中邻近醚 C-O(R)键的 C-O(H)键,其醚选择性大于 72.1%,而且还可广泛用于具有不同取代基的各种其他底物。这项工作表明,透彻了解结构与性能之间的关系对于合理设计和构建具有定制位点的合适催化剂以实现所需的催化反应至关重要。相关成果以“Intermetallic synergy in platinum–cobalt electrocatalysts for selective C–O bond cleavage”为题发表在《Nature Catalysis》上,通讯作者为韩布兴院士和孟庆磊博士,第一作者为Ruizhi Wu。

Pt-Co催化剂的设计与结构表征

图1:电子显微镜表征
为了深入了解Pt-Co金属位点的结构特征,作者使用X射线光谱技术系统地表征了合成的Pt-Co/MC催化剂的精细结构(图2a)。X射线衍射(XRD)图谱显示,与MC载体相比,Pt+Co1/MC催化剂未观察到明显的Pt或Co特征峰,表明其结晶度较差。相反,PtCo1/MC和PtCo/MC催化剂则表现出明确的Pt金属衍射峰,并且这些峰向更高的衍射角位移,显示出增强的Pt-Co金属间相互作用。X射线光电子能谱(XPS)分析进一步揭示了这些催化剂的电子结构(图2b,c),Pt 4f结合能呈现负位移,而Co 2p轨道峰的结合能也相应变化,支持了Pt-Co的良好合金结构。这种相互作用通过Pt L3和Co K边缘的X射线吸收近边缘结构(XANES)曲线进一步证实构中从Co到Pt的电子转移(图2d,e)。
扩展X射线吸收精细结构(EXAFS)分析结果进一步支持了Pt和Co物种之间的金属间相互作用(图2f,g)。定量EXAFS曲线拟合揭示了Pt和Co的配位环境,其中Pt-Co配位数在PtCo1/MC和PtCo/MC催化剂中分别为0.53和1.79,Co的平均配位数为1.10和3.55(图2f)。此外,X射线光电子能谱(XPS)和X射线吸收近边缘结构(XANES)分析表明,PtCo1/MC和PtCo/MC催化剂中的Co原子氧化态较低(图2i),这与Pt和Co之间的电子转移有关。X射线发射光谱(XES)进一步证实了金属间相互作用对Pt和Co原子电荷分布的影响。

电催化性能
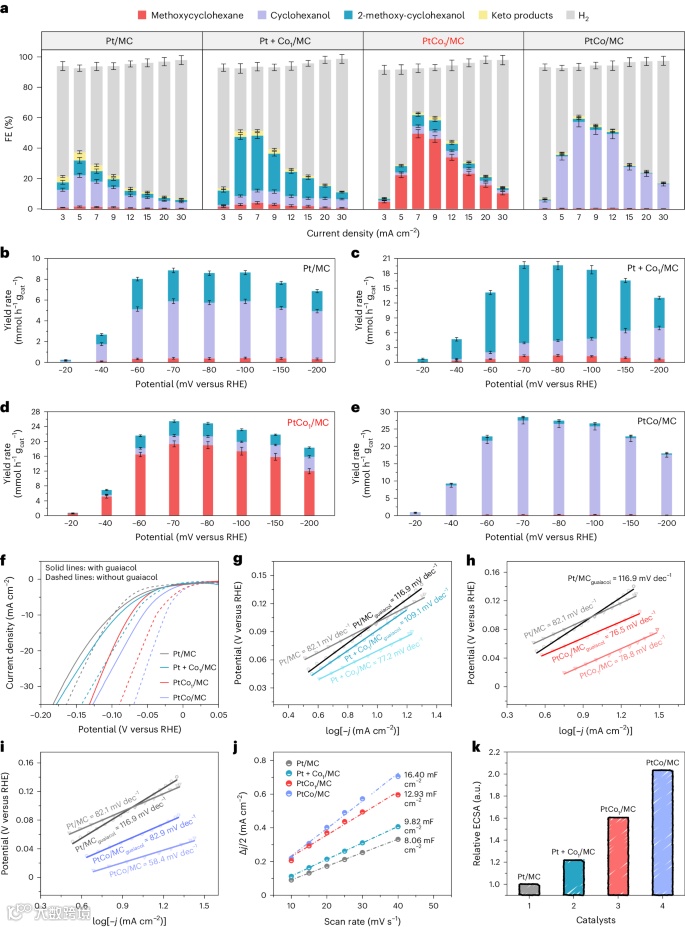
机理探索
为了充分理解PtCo1/MC及其参比催化剂的结构-性能关系,作者通过原位拉曼光谱在最佳条件下监测了愈创木酚的电催化反应过程(4a–f)。拉曼光谱显示,在关闭电源时,反应系统出现了与MC载体相关的特征峰,而在开启电源后,S–O–H伸缩振动峰减弱,反应物的OH–C=C–OCH3伸缩振动增强,表明HSO4−阴离子被愈创木酚分子取代。随着反应进行,HSO4−阴离子和水生成的质子分别参与了芳香环的活化和氢化,表现为相关峰强度的变化和红移。实验还表明,催化剂的存在对于这些拉曼信号的检测至关重要。Pt1+Co1/MC催化剂显示出显著的C–O(H)、(C)O–H和C–O(CH3)信号,而PtCo1/MC催化剂上则C–O(CH3)吸附峰减弱,C–O(H)和芳香π键振动增强,表明愈创木酚的高效HDO反应。此外,电催化脱氧主要依赖于水生成的质子,进一步验证了愈创木酚在PtCo催化剂上的电催化性能。这些结果展示了Pt-Co金属间协作在调控C–O(H)和C–O(CH3)键断裂中的重要性。

图4:原位拉曼研究
作者进行了密度泛函理论(DFT)计算,研究了愈创木酚电催化HDO反应路径,并比较了PtCo1/MC催化剂与单金属Pt/MC和其他两种双金属Pt-Co催化剂(Pt+Co1/MC和PtCo/MC)的反应能量学和吸附方式。DFT计算验证了在PtCo1/MC催化剂上,C–O(H)键的断裂和C–O(CH3)键的保留是最有利的HDO反应路径。反应首先通过氢化芳环,形成中间体2-甲氧基环己-1-烯醇(M4),然后C–O(H)键在较低的能垒下被有效激活,最终生成甲氧基环己烷。Bader电荷分析显示,当2-甲氧基环己-1-烯醇吸附在PtCo1位点上时,Co单原子携带更多电子,揭示了C–O(H)键的电子重新分布和Co-O(H)配位的强相互作用,从而促进了C–O(H)键的断裂。相比之下,C–O(CH3)键的活性较低且对Co SAS不敏感,这进一步确认了Co SAS在弱化C–O(H)键方面的关键作用。通过这种方式,PtCo1/MC催化剂实现了芳香环的氢化和C–O键的选择性断裂。

图5:机理研究
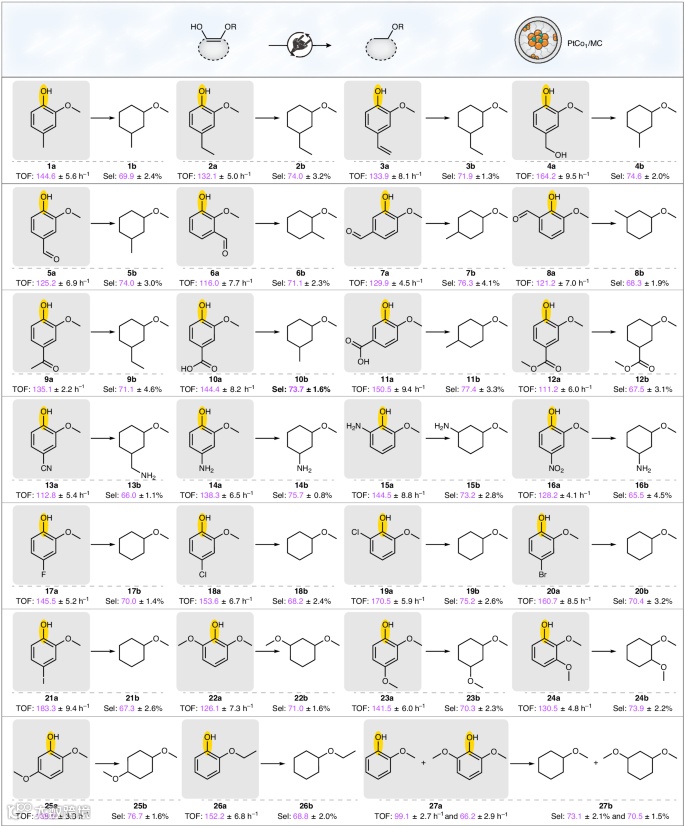
通用性研究
为了表征PtCo1/MC催化剂的加氢和脱氧行为,作者研究了各种取代基(如烷基、烯基、羟甲基、酰基、羧基、酯、氰基、氨基、硝基和卤素)的愈创木酚、二甲氧基酚和2-乙氧基苯酚的HDO反应。结果显示,这些底物在电催化系统中表现出高选择性和高周转频率(TOF),如烷基和烯基取代的愈创木酚产物选择性超过69.9%,TOF范围为132.1-144.6 h⁻¹;羟甲基、酰基、羧基和酯基取代的愈创木酚产物选择性在67.5-77.4%,TOF为111.2-164.2 h⁻¹;氰基取代的愈创木酚在TOF为112.8 h⁻¹下生成保留C–O(CH3)键的产物。卤素取代的愈创木酚选择性约为70.0%。这些结果表明,PtCo1/MC催化剂在断裂C–O(H)键和保留C–O(CH3)键方面具有高效性和广泛兼容性。
声明

“邃瞳科学云”直播服务

扫描二维码下载
邃瞳科学云APP



