


理解材料的构-效关系一直是电池领域的关键课题。随着先进表征技术的发展,研究人员能够更加准确、便捷地获得材料的物理化学性质,从而促进高效、稳定的电池材料的开发。美国easyXAFS公司设计研发了台式X射线吸收精细结构谱仪easyXAFS300+,该装置无需同步辐射光源,可以在常规实验室环境中实现X射线吸精细结构谱(XAFS)和X射线发射谱(XES)双功能测试,获得媲美同步辐射光源数据质量的谱图。近年来,不少研究者利用easyXAFS300+系统实现了对元素化学价态、局部配位结构以及自旋态的多重互补信息的获取,为阐明电化学性能的改善机理提供了关键数据支撑。

图1. easyXAFS台式X射线吸收精细结构谱仪

本文将从ACS Mater. Au, ACS Appl. Mater. Inter., Angew和JACS等代表性平文献出发,阐述easyXAFS台式X射线吸收谱仪系统如何助力电池材料机理研究。
Maximilian Fichtn课题组合成了锰基无序岩盐氧氟化物Li2Mn1−xVxO2F(0≤x≤0.5)作为锂离子电池正极材料,并利用easyXAFS台式X射线吸收精细结构谱仪的easyXAFS300+确定合成材料中Mn和V的氧化状态[1]。如图2a所示,原始的LMOF、以及经20个循环的LMOF和HT-LMOF XANES光谱与Mn2O3的吸收边能量一致。这表明Mn3+处于放电/锂化状态,与原始LMOF和前20个循环的锰可逆的氧化还原反应类似。如图2b所示,虽然两个循环的电极都表现出比原始材料更高的振幅,但扩展边(EXAFS)数据的傅里叶变换证实了他们相同的局域配位,这表明循环后局部无序化降低。在HT-LMOF_20C中,观察到第一和第二配位壳的傅里叶变换峰振幅略高,这表明热处理减少了局部无序化现象。对第一个Mn-O/F和第二个Mn-Mn配位壳层进行拟合。对于HT-LMOF来说,Mn-O/F的原子间距离变大,Mn-Mn的配位键长略有增加。可以推断,热处理有助于提高球磨化合物的对称性并减少缺陷,但也可能影响结构中的局部氟化程度。
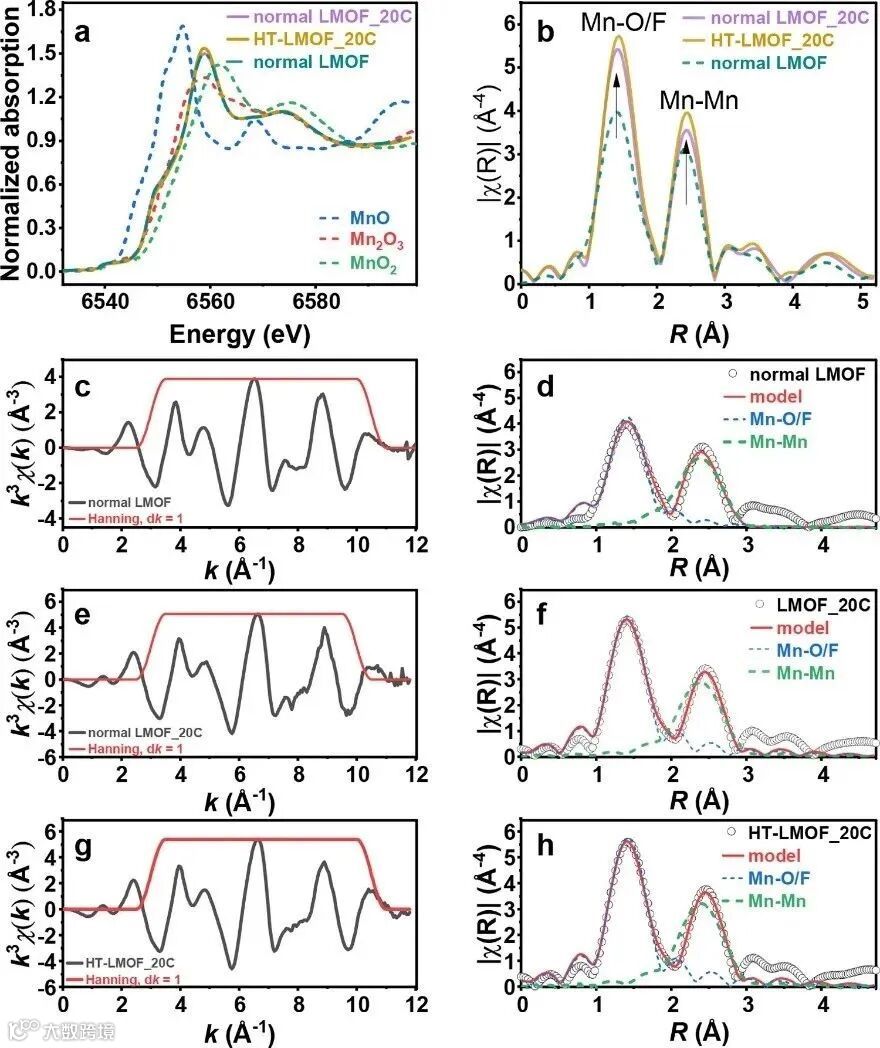
图2. 原始LMOF、以及LMOF及HT LMOF 20个循环后的Mn k边XAFS光谱。(a)标准物的XANES; (b) EXAFS的傅里叶变换,原始LMOF (c和d)、原始LMOF_20C (e和f)和HT-LMOF_20C (g和h) 的R空间壳层拟合;k3加权χ(k), dk = 1。

图3. 放电状态下,LMVOF_20C电极的 (a) V和 (b) Mn 的K边XANES光谱。
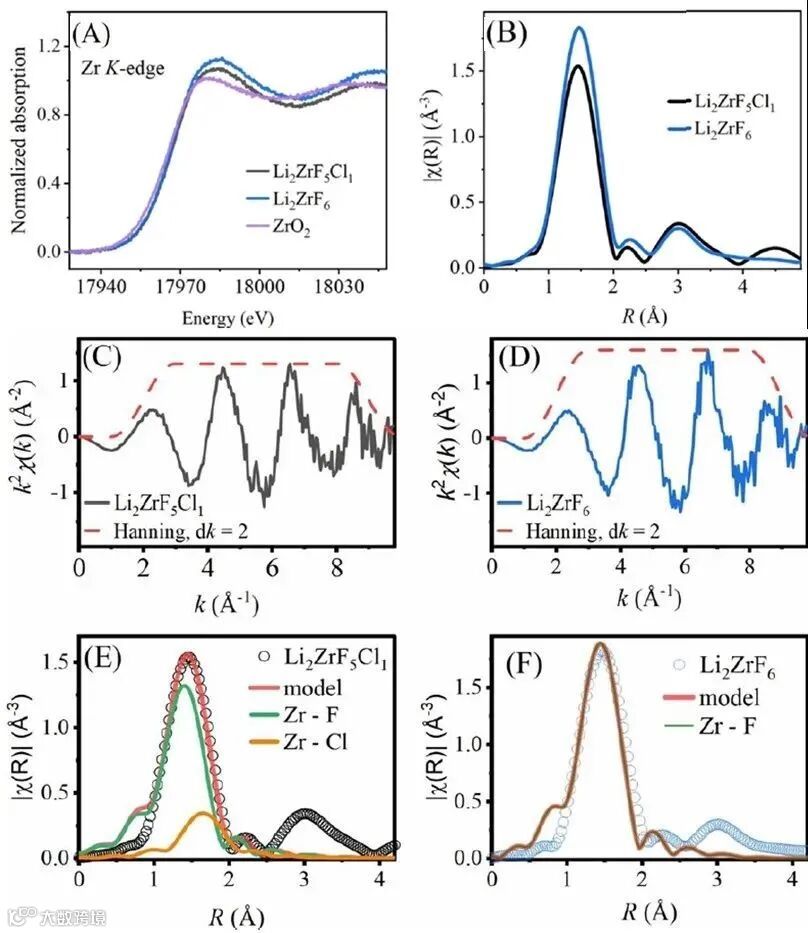
图4. (A) 归一化的Li2ZrF6,Li2ZrF5Cl1 及ZrO的Zr XANES 谱图; (B) k2 加权 χ(k) 的傅立叶变换; (C, D) k2 加权 χ(k);(E, F)第一配位层的拟合 (1~2.5 Å)。

苏州大学张亮团队针对Li-S电池反应体系,以尖晶石氧化物为基础,构筑了吸附-催化双活性位点,并通过自旋调控形成电荷传输通道[3]。作者成功合成了富含MnOh-O-CoTd结构单元的三金属尖晶石氧化物纳米盒(CoFeMnO YSNCs),处于四面体中心的高自旋CoTd以金属-硫共价键的形式强力地锚定多硫化物,处于八面体中心的MnOh能诱发轨道特异性催化。同时,利用金属离子在特定配位构型中的电子结构及自旋态,MnOh-O-CoTd中形成了畅通的自旋路径,保证了双活性位点之间的电子传递。
作者利用easyXAFS台式X射线吸收精细结构谱仪的XES Kβ谱线验证CoTd的自旋态(图5)。在CoFeO SSNC样品中,Kβ1,3-Kβ′分裂难以区分,证明其中低自旋的八面体中心Co3+Oh占主导地位。与之相比,CoFeMnO YSNC的Kβ′特征增强,证实其中更多的Co3+Oh被驱动到四面体配位中心Td,并转换为高自旋状态。

图5. 不同样品的Co Kβ XES谱图
由于低自旋Co3+Oh的固有局部电子结构与空的eg轨道,具有CoOh-O-CoTd结构单元的Co3O4显示出相对较低的电导率(图6a)。相反,当Mn3+被引入Oh位点时,Mn3+Oh中部分占据的eg轨道通过桥接O2-带来强烈的MnOh-O-CoTd超交换相互作用,构建了一个畅通无阻的自旋通道(图6b)。如图6c所示,自旋极化电子在费米能级附近具有自旋态,通过离域电子表现出半金属特性。双活性位点间通过自旋极化电子形成有效的电荷传输通道,促进多硫化锂在电极表面发生连续的吸附-催化转化过程(图6d),实现了高性能的Li-S电池。该项研究以“Cooperative Catalysis of Polysulfides in Lithium–Sulfur Batteries through Adsorption Competition by Tuning Cationic Geometric Configuration of Dual-active Sites in Spinel Oxides”为题,发表于国际高水平期刊《Angewandte Chemie International Edition》。

图6. (a) Co3+Oh和相应的CoOh-O-CoTd自旋通道的轨道分裂图示。(b) Mn3+Oh和相应的MnOh-O-CoTd自旋通道的轨道分裂图示。(c) MnOh掺杂Co3O4的自旋极化示意图。(d) 多硫化锂在CoFeMnO YSNCs表面连续“吸附-转化”过程的机理示意图。

中国科学院的刘向峰团队利用自旋态调控设计出稳定的4.6V高压LiCoO2正极材料,有效提升了电池容量和倍率性能,并实现了优异的充放电循环稳定性[4]。高压LiCoO2由于其较高的理论容量而备受研究者关注,然而高电压会导致过量的O→Co电荷转移,使晶格O2–过度氧化并释放O2,造成结构退化,容量衰减,严重限制其实际应用。本文的作者通过引入高自旋的Co有效解决了这一问题,其机理如图7a所示。对于低自旋LiCoO2而言,Co t2g和O 2p轨道之间高度重叠。充电后,首先从t2g中提取电子,将低自旋Co3+(t2g6 eg0)氧化为Co4+(t2g5 eg0)。当一半的Co3+阳离子被氧化成Co4+时,费米能级达到O 2p态。随着进一步氧化,O→Co电荷转移被触发,从而将Co4+还原为Co3+甚至Co0+,同时将O2−氧化为O2。这一过程使O-O 键缩短,Co-O键变长,造成晶格畸变。充放电循环后,由于不可逆的化学反应和晶格变化,材料的电子结构无法恢复到原始状态。对于高自旋LiCoO2,Co t2g和O 2p之间的重叠减少。纳米带状网络结构产生的晶格应力场抑制了脱锂时的结构变化。这些特性抑制了费米能级在高电压下接近O 2p,并从根本上解决了O → Co电荷转移问题。因此,充放电循环后,高自旋LiCoO2的能带结构可恢复到初始状态。
实验上,作者成功合成出高自旋LiCoO2 (HS-LCO)与低自旋LiCoO2(LS-LCO),并利用easyXAFS台式X射线吸收精细结构谱仪的XES辅助表征(图67b)。在长时间电池循环测试后,HS-LCO中的Co仍然保持高自旋态。该项研究以“Reducing Co/O Band Overlap through Spin State Modulation for Stabilized High Capability of 4.6 V LiCoO2”为题,发表在国际顶级期刊《Journal of the American Chemical Society》上。

图7. (a) LS-LCO和HS-LCO中自旋态对能带结构的影响及氧化还原机理示意图。(b) LS-LCO和HS-LCO原始样品及循环测试后的HS-LCO电极的XES谱图。

来自美国华盛顿大学XAFS科学家多年的技术积累,理论和技术底蕴雄厚,设备成熟稳定;具有XAFS和XES两种工作模式,可快速切换,满足不同科研试验需求;
台式设计,可以在实验室内随时满足日常样品分析。
设备运行超稳定,操作便捷,维护成本超低,安全可靠;
全自动软件控制,可以同时进行多个样品或样品参数条件下的测试;
台式XAFS/XES谱仪测得的谱图效果与同步辐射数据一致,如下图所示,其测得的Ni元素的EXAFS,Ce和U元素的L3-edge的XANES谱图数据与同步辐射光源谱图效果完全一致;


用户友好软件界面,全自动软件控制测试过程,可以同时进行多个样品或样品参数条件下的测试配置灵活,适用于对空气敏感的样品的检测或一些原位测试,如原位的锂电池或电催化实验测试,监测电极/催化材料的结构变化;
多种型号和配置可选,满足不同科研要求

【参考文献】
[1] Cooperative Catalysis of Polysulfides in Lithium–Sulfur Batteries through Adsorption Competition by Tuning Cationic Geometric Configuration of Dual-active Sites in Spinel Oxides. Angew. Chem. Int. Ed., DOI: https://doi.org/10.1002/anie.202216286
[2] Reducing Co/O Band Overlap through Spin State Modulation for Stabilized High Capability of 4.6 V LiCoO2. J. Am. Chem. Soc., DOI: https://doi.org/10.1021/jacs.3c01128


微信号:QuantumDesign
扫码关注获取更多内容
Quantum Design,源于科学,致力科学!

戳“阅读原文”即刻咨询easyXAFS吧!