


第一作者:闵媛博士
通讯作者:陈洁洁教授、俞汉青教授、熊宇杰教授
通讯单位:中国科学技术大学
论文连接:https://www.nature.com/articles/s41467-023-40906-6

碳卤键的活化在环境和能源领域具有重要意义。一方面,以碳卤键断裂为关键步骤,可以提升卤代有机污染物的可生物利用性,有助于改善传统水处理效果,促进卤素的地球化学良性循环;另一方面,卤代有机物作为有机合成中非常重要的合成前体,广泛用于偶联、加成、还原性转化等反应。其中,有机氯化物的强碳氯(C-Cl)键(327 kJ mol-1)使其表现出化学惰性,具有易低成本且易于储存等优点,因此可将其作为一种耐受性良好的后期官能团衍生化方式。相比于有机相体系,水相中有机氯化物的C-Cl键活化机制仍没有得到充分探索。其中,溶剂水分子对C-Cl键的还原活化具有一定的抑制作用。尽管如此,水中的化学转化与合成反应具有安全和可持续的特点,正成为更绿色的关键赋能技术。因此,在水中卤代有机物的污染治理与高附加值化学品的合成转化等应用领域,亟需发展高效的碳卤键活化策略。
电催化还原反应以多重质子耦合电子转移的方式来转化并去除污染物。其中,还原脱氯反应可在温和条件下实现氯代有机污染物碳氯键的活化与断裂。然而,设计具有高活性和高选择性的电催化剂,仍是一个颇具挑战性的任务。从反应位点的原子结构方面,一种可行的设计思路是调控金属中心和配位结构;从位点附近的微环境方面,通过构建不同空间层次的电催化剂,可充分利用最新发展的功能材料,包括维度由低到高的分子催化剂、量子点、纳米颗粒、纳米线、纳米片和多孔结构等。因此,从反应位点和微环境两方面对电催化剂进行设计,不仅要求对电催化反应机理有深入的分析与认识,也需要对电极界面过程有精确的调控策略。
天然酶为能源与环境领域的催化剂发展提供了设计灵感。其中,金属酶可作为研发小分子过渡金属催化剂的起点,以实现类似于生物体系的化学催化活性。作为一类能够活化碳卤键的钴分子催化剂——天然存在的还原脱卤酶的B12辅因子,为设计电催化的活性位点结构提供了参考。除了钴催化活性中心,B12周围肽链形成的内部空间和动态结构对反应选择性也具有重要作用。例如,能够锚定并活化底物或过渡状态以形成目标产物。然而,大多数催化剂结构通常较为刚性和静态。因此,在电极表面构建一个类酶化动态环境将有利于强化电催化活性与选择性。同时,在电极-电解质界面过程中空间效应作用的明晰也将为催化剂的设计提供理论支撑。
针对上述目标,该工作以不依赖于化学键,也不受限于材料晶格匹配度的范德华异质结为动态可控界面电催化剂设计平台,将B12的高还原活性与二维材料的物质及电荷传输性质相耦合,构建了仿脱卤酶电催化体系。相较于仅有外部催化表面的传统块材电极,范德华异质结电极实现了外表面与亚表层的双重催化特性,从而显著增强了氯代有机污染物脱氯能力。通过电化学分析、理论模拟和原位光谱等手段,分析了污染物在范德华异质结层间锚定行为,揭示了层间空间的去溶剂效应使污染物的还原电位正移,从而使其更易于接受电子并发生还原脱氯的过程。该工作通过学习天然酶的催化机制,发展了将电催化剂活性位点与动态内部空间结构设计集成的策略,为复杂条件下的电催化界面过程优化提供了新思路。

2)该工作揭示了通过层间空间的去溶剂效应,促使水中污染物的还原电位正移,从而使化学惰性的碳卤键易于接受电子,实现高效的还原加氢反应。

为设计和组装结合袋状的电催化剂,对B12依赖性脱卤酶的结构和功能进行了全面剖析。微生物还原脱氯发生在脱卤酶的结合袋处,包括辅因子(B12)和周围带有芳香环和羟基的关键残基,而具有C−Cl键的底物分子是微生物有机卤化物呼吸的最终电子受体。与水中的均相催化不同,脱卤酶的疏水内部空间为传递质子的狭窄通道提供了局部环境,而受限的水分子能够稳定与周围芳香残基的B12−CONH2和-OH基团之间形成不连续的氢键网络。为探讨B12分子催化剂的作用以及内部空间对还原脱氯的空间效应,构建了三种阴极结构的电化学系统:不含B12的GO电极、表面吸附B12的GO-B12电极和带有额外嵌入B12的GO-B12-GO电极。采用MD模拟分析电极组装过程,得到层间距离和分解能量的变化曲线,发现GO的堆叠行为是由范德华力、B12-CONH2的氢键和水的溶剂化作用驱动。此外,通过同步辐射光电子能谱(SRPES)分析电极的元素深度分布情况,证实了不同的层状结构。X射线吸收精细结构(XAFS)光谱用于分析GO-B12-GO的R空间,在1.88 Å处出现了Co-N配位峰,没有发现Co-Co贡献的特征峰。且电子能量损失谱(EELS)和高分辨率X射线光电子能谱(XPS)分析显示C、N和Co元素均匀分布。
此外,通过多次扫描循环伏安法(CV)测试,发现GO-B12-GO电极的电化学活性面积大约是GO-B12电极的3.8倍,比单独的B12电极高27倍。因此,GO-B12层间空间较小导致内表面有限,而GO-B12-GO的层间空间较大导致其内表面较大。紫外可见吸收光谱在360 nm处出现B12分子的特征峰,定量分析表明GO-B12-GO的负载量为0.0576 mg cm-2,而GO-B12(0.0365 mg cm-2)或单独的B12(0.0363 mg cm-2)的含量较低,表明GO-B12-GO的额外负载量归因于其嵌入内部空间。电容分析结果表明,GO-B12-GO电极的层间距离大于GO,后者的扩散贡献较高(93%),这归因于Na+离子与GO电极内表面之间的距离较小。GIXRD图谱也证实了这一点,GO电极在10.047°处有衍射峰,对应于0.880 nm的层间距离,而GO-B12-GO电极的层间距离较大(1.088 nm)。此外,小角X射线散射(SAXS)图案在方位角≥90°处显示出明确的各向同性散射,表明GO-B12-GO电极在电化学处理后具备了层状排列的结构。

图1仿酶层状电极构建
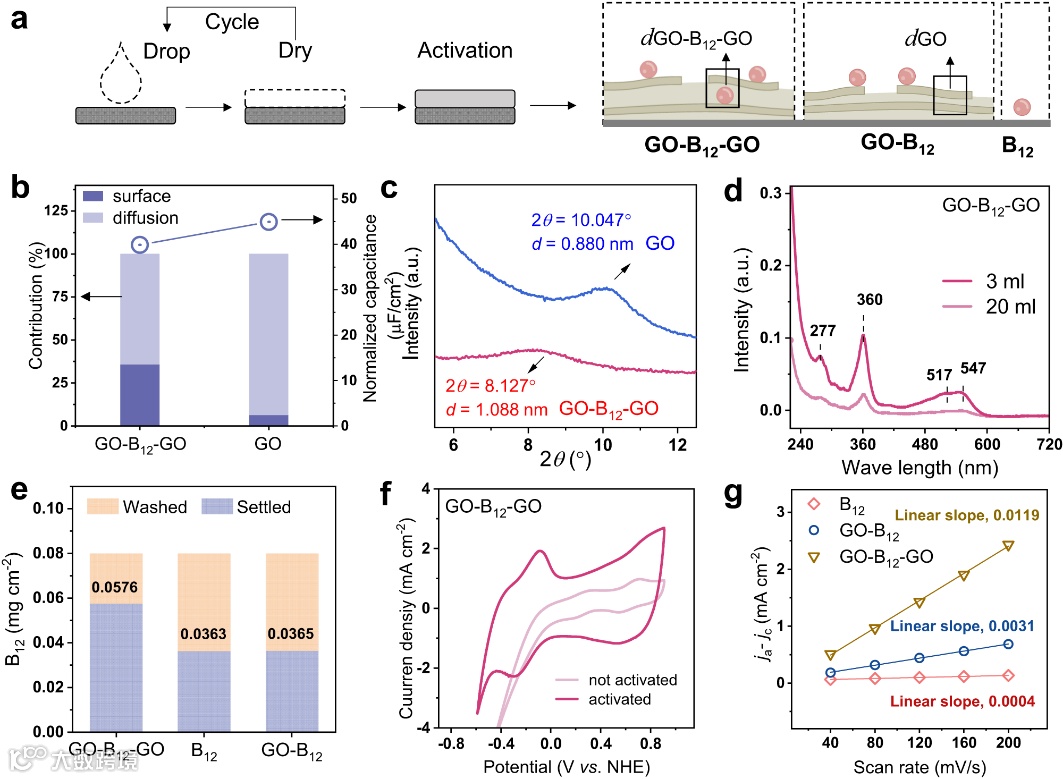
图2 电化学活化处理
以三氯乙酸钠(TCA)为探针分子,对电催化还原能力进行了分析。与空白电解液相比,加入TCA后电流密度增加,说明GO-B12-GO电极具有还原TCA的能力。然而,GO电极在相同的条件下的电流密度并无增加,表明GO电极没有催化活性。为了增强GO-B12-GO层状电极的还原能力,可通过增加B12使其形成一个从溶液指向电极的浓度梯度,从而扩散至电极表面发挥作用。但实验表明,TCA的还原电流密度并没有随着B12的增多而提升,松散吸附在电极上的B12的贡献可忽略不计。进一步,构建其他层状电极(GN-B12-GN)也验证了B12分子的空间分布对电化学催化的作用。因此,层状电极上的结合态B12发挥了重要作用,包括外表面的B12以及内部空间的B12。
基于上述分析结果,采用恒电位测试比较了不同电极的性能,并对TCA电还原反应中的氯元素进行了衡算。GO电极的TCA还原转化率最低(17%),而含有B12的电极表现出更高的还原能力,其中GO-B12-GO电极实现了86%的转化率。有趣的是,对于检测到的中间体DCA,单独GO、B12或GO-B12电极上的DCA浓度在3 h内不断累积(TCA → DCA),而GO-B12-GO电极在1 h时出现了一个DCA浓度拐点,表明DCA先少量累积,但快速消耗并转化为MCA。对于DCA还原反应,GO-B12-GO的表观速率常数(k2=1.49 h−1)分别是GO-B12和B12的7.8和5.7倍。为了进一步理解GO-B12-GO电极为何表现出更高性能,假设GO-B12和GO-B12-GO电极上B12的催化活性相同,根据B12-GO电极的DCA转化活性,计算出GO-B12-GO电极实验性能所需的B12理论数量为1.54 × 10−7 mol cm−2,远大于观测到的B12负载量(4.25 × 10−8 mol cm−2)。此外,衰减全反射傅里叶变换红外光谱(ATR-FTIR)的电化学原位测试也验证了层间B12在促进电化学还原方面发挥着关键作用。因此,GO-B12-GO电极中插层的B12比表面吸附的B12具有显著更高的活性。
为了验证这种层状电极优势的普适性,采用钴酞菁(CoPc)作为替代的钴中心催化剂。GO-CoPc-GO电极对TCA转化效率达到80%(3 h),而GO-CoPc电极的转换效率显著降低了20%,证实了这种电极结构层间空间对电化学还原的促进作用。然而,值得指出的是,与GO-B12-GO相比,GO-CoPc-GO对DCA向MCA的还原转化有限,导致了DCA积累(TCA→DCA)。为了探讨不同钴分子催化剂之间的性能差异,也采用了DFT计算来比较B12和CoPc的本征活性,从轴向配体(5,6-二甲基苯并咪唑)角度补充分析了其对C−Cl键断裂的影响。

为理解插层B12与层间空间对DCA还原反应的促进作用,进行了MD模拟。在初始状态,所有DCA离子位于远离GO-B12-GO电极的溶液中。随后,DCA在溶剂和电极的非共价相互作用下向四周扩散。体系达到平衡状态时,其空间分布如下:一个DCA吸附在GO表面,两个DCA分别进入GO的层间空间,另一个仍在水中游离。其径向分布函数g(r)显示第一个窄峰,随后是两个宽峰,表明DCA和水分子之间的距离(DCA−OW)分别为0.28、0.36和0.49 nm。因此,DCA的平均配位水分子数(CN)从高到低分别为:在溶液中的(CN = 6.5)、吸附在表面上的(CN = 4.8)与嵌入内部空间的(CN = 4.1、3.4)。其中,CN值是在DCA-OW距离为0.42 nm的第二个溶剂化壳层内计算,运行时间延长至50 ns。在插层DCA周围发现了不太紧密的溶剂化结构,这归因于GO和B12的静电和氢键相互作用,形成了类似于酶结合口袋的局部环境。在本体溶液中,由于实验pKa为1.2639,DCA被去质子化,从而为脱氯反应提供了0.870 eV的能垒(DCA → MCA)。相比之下,层间空间中的DCA部分被溶剂化,提供了改变质子化状态的机会,这将有助于通过较低势垒脱氯。因此,脱氯反应途径可以通过氯化有机污染物周围的溶剂化结构来调节。
为了模拟大量水中的氢键网络,DCA·nH2O在没有结构限制的情况下被弛豫到最低能量状态,发现带隙随着水数的增加而增加的趋势。由于DCA周围水的配位结构处于动态平衡,实际构象空间远大于上述最低能量构象。因此,根据MD模拟结果(CN = 6.5),发现相应的溶液中DCA(n = 5–8)具有的导带底能级(CBM)为–2.0 eV。相比之下,气相的DCA(n = 0)显示出最低的费米能级为-4.844 eV,CBM为-2.467 eV,这有利于从外部电极接受电子。在部分去溶剂化状态下(n = 1–4),CBM的范围为-2.409至-1.937 eV。结合分子轨道分析,最低未占据分子轨道(LUMO)由DCA的C−Cl键贡献,而具有较低空轨道的DCA将更容易接收电子、发生C−Cl键的还原反应。
除了最低能量结构外,不连续氢键网络的空间应变增加的能量可以调节DCA的去溶剂化结构。为了研究电极配置对溶剂化结构和氢键网络的阻碍作用,以n = 3为例,具有四种可能的状态(S1至S4)。随着形成能较高(S2至S4),DCA·3H2O变得不太稳定。与此同时,LUMO能级显示出从-1.918到-2.473 eV的负移,表明电子的接受更有利。此外,OH的距离从1.251减少到1.051 Å,导致从去质子化(COO-)到质子化(COOH)的状态变化。去质子化的DCA (S1)显示通过大量已知的Grotthuss机制进行的连续质子扩散,具体取决于水合氢离子和水之间的相互作用。相反,质子化的DCA(S2至S4)由于离散的氢键网络而显示出保存完好的O−H键,这将有利于以较低的能垒进行还原脱氯。
因此,GO-B12-GO层状电极通过空间效应调控质子化状态、最小化所需的过电势,实现了电化学催化还原的性能提升。为了实现更多小底物的还原,进一步将目标底物的反应性通过其分子轨道和电化学势的关系来评估。对于氯乙酸,LUMO能级的顺序为MCA>DCA>TCA,这与实验转换效率的顺序一致。与该反应规律一致,硝基苯是一种比氯乙酸盐更容易进行电化学还原的已知底物,其表现出的LUMO能级要低得多;还考察了氯酚和氯乙烷等其他有机底物,为电化学还原的难度提供了参考基础。此外,底物和层间空间的尺寸匹配可能在去溶剂化程度和进一步提高转化效率中发挥重要作用。



该工作基于分子催化剂(B12)和2D材料(GO)的仿酶结合袋状电极能够提供内部空间以促进C−Cl键的电化学还原。其中,插层有机底物分子通过不连续的氢键网络改变质子化状态,去溶剂化效应能够最小化所需的还原反应过电势,从而实现碳卤键的还原活化。因此,该研究表明除了表面催化过程,电极的内部空间也可用于调节催化还原性能。鉴于可用的分子催化剂种类多样,精确的活性位点与动态内部空间的集成是许多电化学催化反应的适用策略。同时,通过利用新兴的二维材料和多孔材料,为设计多层次结构的仿酶复合电极提供了新思路,对构建用于环境与能源等领域的高性能电催化剂提供了技术支撑。
该研究工作得到国家重点研发计划和国家自然科学基金的支持,相关研究结果近期发表于Nature Communications期刊。中国科学技术大学博士后闵媛为论文的第一作者,陈洁洁教授、俞汉青教授和熊宇杰教授为论文的共同通讯作者。

闵媛,中国科学技术大学博士后,2022年于中国科学技术大学获博士学位。主要从事酶催化机制解析、仿生电催化剂设计与环境污染物降解性能调控等方面的研究工作,以第一作者身份在Nature Communications上发表2篇论文,以共一作者在ACS Nano和Water Research等SCI刊物发表4篇论文。

陈洁洁,中国科学技术大学教授。国家优青获得者。2014年于中国科学技术大学获得环境工程专业博士学位,并继续从事博士后研究工作,2015-2016年赴美国加州大学-伯克利分校科学院院士Graham Fleming教授课题组访问交流。主要从事污染物的生物与化学转化研究工作,在污染物转化中的界面电子转移机制解析、调控方法、系统构建等方面进行了研究。以第一/通讯作者在Nature Communications、PNAS、Advanced Materials、Environmental Science & Technology、Water Research等SCI 刊物发表论文30余篇。
俞汉青,中国科学技术大学环境科学与工程系教授。1994年在同济大学环境工程学院获博士学位后,先后在境外三所大学从事研究工作,2001年获中国科学院人才计划回国工作,为国家自然科学基金委杰出青年基金获得者。2014年以来连续入围 Elsevier 出版集团环境领域国际高被引学者,科睿瑞安(Web of Science)交叉领域的高被引科学家。主要从事水污染控制的基础研究、技术研发和实际应用工作,获授权发明专利60多项,部分获实际应用。作为通讯作者在包括Nature子刊、PNAS、Environmental Science & Technology和Water Research在内的SCI刊物发表论文500多篇,SCI他引超过4万次,H因子109 (Web of Science);成果获国家自然科学二等奖和国家科技进步二等奖各1项,以及省部级科技/自然科学一等奖7项。
熊宇杰,中国科学技术大学讲席教授、博士生导师。1996年进入中国科学技术大学少年班系学习,2000年获化学物理学士学位,2004年获无机化学博士学位,师从谢毅院士。2004至2011年先后在美国华盛顿大学(西雅图)、伊利诺伊大学香槟分校、华盛顿大学圣路易斯分校工作。2011年辞去美国国家纳米技术基础设施组织的首席研究员职位,回到中国科学技术大学任教授,建立独立研究团队。2017年获国家杰出青年科学基金资助,入选英国皇家化学会会士(FRSC)。2018年获聘长江学者特聘教授,入选国家万人计划科技创新领军人才。2022年当选东盟工程与技术科学院外籍院士(FAAET(F))、新加坡国家化学会会士(FSNIC)。现任ACS Materials Letters副主编。长期从事光电催化研究,通过发展“精准合成-多域表征-数据驱动”交叉研究范式,推动生态系统重构应用。已发表200余篇通讯作者论文(60篇Nat. Commun.、J. Am. Chem. Soc.、Angew. Chem.和Adv. Mater.),入选科睿唯安全球高被引科学家榜单、全球前2%顶尖科学家榜单、爱思唯尔中国高被引学者榜单(引用36,000余次,H指数96)。2012年获国家自然科学二等奖(第三完成人),2014-2016和2018年四次获中国科学院优秀导师奖,2015年获中美化学与化学生物学教授协会杰出教授奖,2019年获英国皇家化学会Chem Soc Rev开拓研究者讲座奖,2021年获安徽省自然科学一等奖(第一完成人)
声明

“邃瞳科学云”直播服务

扫描二维码下载
邃瞳科学云APP



