


作者:一只小羊
审核:peacestore
Xenium 是 10x Genomics 推出的一个用于空间转录组学的实验平台,能够捕捉单细胞层级的转录本表达信息,并且提供丰富的空间定位数据。下面将详细介绍 Xenium 下机数据的结构、常见的文件格式及其用途,以及如何使用不同工具(如 Xenium Explorer、Python 和 Seurat)进行数据读取和分析。
❐ Xenium 数据下机后的文件 ❐
做xenium分析,我们从公司拿到手的就是以下格式的文件。不过随着版本迭代,文件内容有些许改变。以下是针对xenium ranger (XR) v3.0~v3.1,Xenium Onboard Analysis (XOA) v3.0~v3.3之间的数据。

2025年8月,XOA系统更新为4.0版本,9月XR也同步更新到4.0
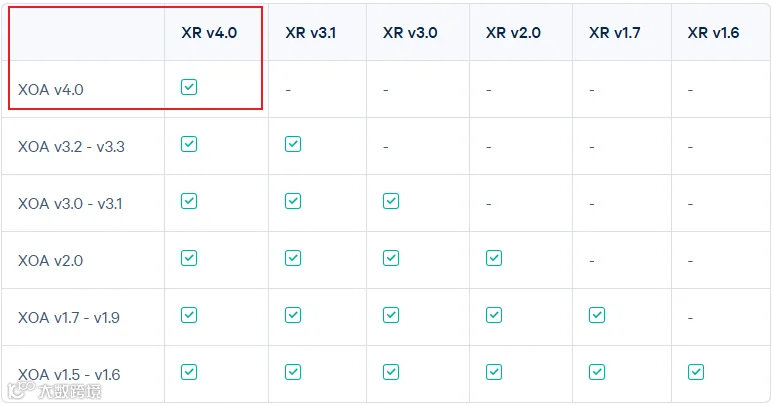
结果的部分内容有所改动,同时单通道荧光图命名也有些许改变,不影响后续分析,但是在使用xenium explorer的时候需要使用v4.0(我们下一篇推送会详细讲解xenium explorer的使用):
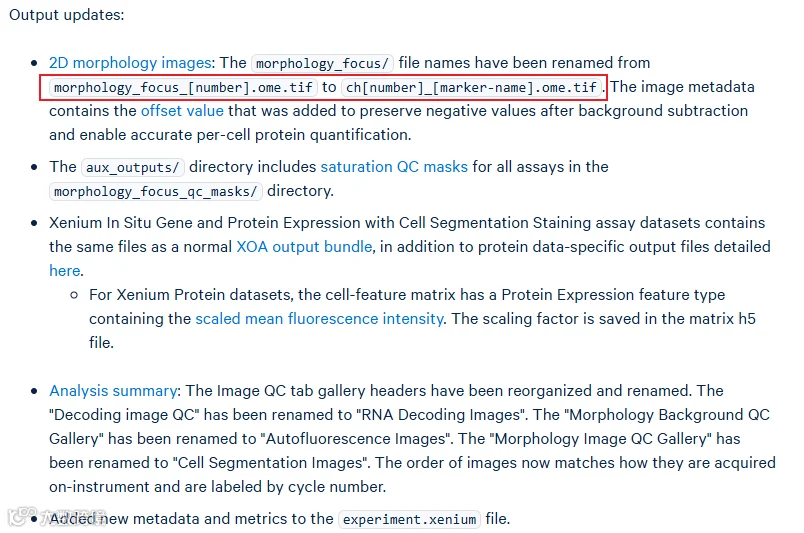
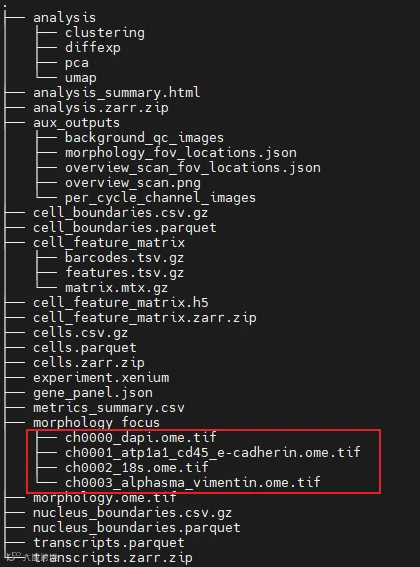
此外,自从2024年6月之后,为了优化读取速度,transcripts.csv.gz就不再出现于结果目录中了,因此导致结果中cells和transcripts的结果文件不太对称。

(1)使用xenium explorer查看数据
使用xenium explorer,至少需要的文件包括:
○ experiment.xenium:作为索引,加载以下对应文件
○ transcripts.zarr.zip:转录本的位置信息
○ cells.zarr.zip:细胞信息的汇总文件,包括细胞/细胞核分割掩码/边界
○ cell_feature_matrix.zarr.zip:用于提供每个细胞中每个基因的计数结果,在圈选区域查看每个细胞中基因表达情况时发挥作用。
○ analysis.zarr.zip:用于提供细胞的分群信息
○ morphology_focus:提供每个荧光通道的图片信息
○ analysis_summary.html:xenium explorer中的菜单栏有设置此信息的加载,所以需要此文件
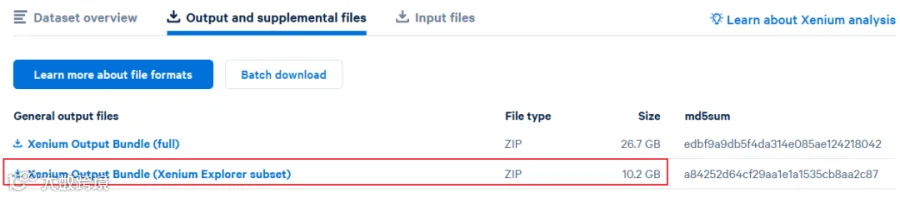
在10x官网下载数据时,有的就会提供这样一个简单版本的数据以供查看:
(2)使用python对xenium数据进行后续分析
python包spatialdata_io可以读取xenium下机文件,具体使用的文件为:
○ experiment.xenium:用于提供重要的版本信息、像素大小等
○ nucleus_boundaries.parquet:提供细胞核的边界信息
○ cell_boundaries.parquet:提供细胞的边界信息
○ transcripts.parquet:提供转录本位置信息
○ cells.parquet:提供cell metadata中的z_level和nucleus_count
○ cells.zarr.zip:用于提供cell metadata信息
○ cell_feature_matrix.h5:表达矩阵
○ morphology_focus:荧光图像
以上信息读取之后得到的数据为sdata,格式如下:
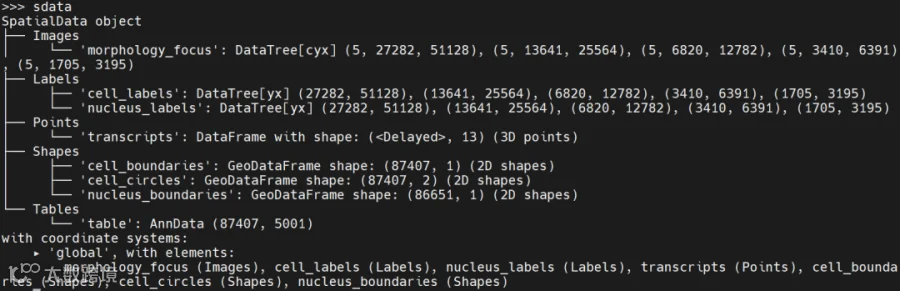
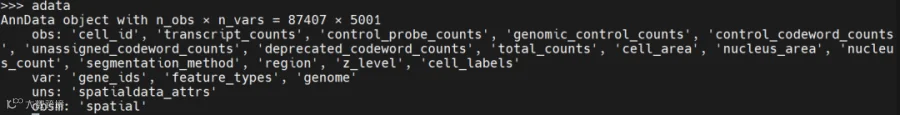
其中table就是表达矩阵,取出来可以看到以下信息:
分析代码:
import scanpy as sc
from spatialdata_io import xeniumsdata = xenium(xenium_path)# xenium_path就是前面展示的目录结构的地址adata = sdata.tables["table"]sc.pp.filter_cells(adata, min_counts=10)sc.pp.filter_genes(adata, min_cells=5)adata.layers["counts"] = adata.X.copy()sc.pp.normalize_total(adata, inplace=True)sc.pp.log1p(adata)sc.pp.pca(adata)sc.pp.neighbors(adata)sc.tl.umap(adata)sc.tl.leiden(adata)
(3)使用R对xenium数据进行后续分析
R包seurat中的LoadXenium函数可以读取xenium数据,需要的信息基本和spatialdata_io中的函数一致,不过读取的文件是h5/parquet/csv.gz,优先读取parquet,如果没有则读取csv.gz格式的文件,最大的区别是不会读取图片信息。
需要注意的是,seurat从v5.2.0开始才针对下机文件中没有transcripts.csv.gz进行了修改。所以如果seurat版本较早,那么会出现读取问题,报错没有此文件。
分析代码:
library(Seurat)
options(future.globals.maxSize = 10 * 1024^3) # 设置内存限制为 10 GB,如果不够需要再调大
# Load the Xenium data
xenium.obj <- LoadXenium(path, fov = "fov", segmentations = "cell")
# remove cells with 0 counts
xenium.obj <- subset(xenium.obj, subset = nCount_Xenium > 0)
xenium.obj <- SCTransform(xenium.obj, assay = "Xenium")
xenium.obj <- RunPCA(xenium.obj, npcs = 30, features = rownames(xenium.obj))
xenium.obj <- RunUMAP(xenium.obj, dims = 1:30)
xenium.obj <- FindNeighbors(xenium.obj, reduction = "pca", dims = 1:30)
xenium.obj <- FindClusters(xenium.obj, resolution = 0.3)
Xenium 下机数据每种格式文件用途汇总
✧ Zarr 格式:主要用于 Xenium Explorer 中的数据加载。Zarr 格式数据具有分块存储和读取、高效压缩、并行访问的特点,适用于存储大规模高维数据,如转录本的位置信息、细胞信息、分析结果和图像数据等。Zarr 文件结构类似于一个层级化的目录系统(类似于 HDF5),每个目录下可以存储不同的数组数据,文件内部的组织结构非常灵活。
✧ Parquet 格式:用于 Python 和 R 读取分析。Parquet 格式适用于结构化数据,如细胞元数据、转录本的位置信息和基因表达矩阵等。相比 Zarr 格式,Parquet 格式更适合进行快速的列式查询。
✧ CSV.gz 格式:相当于是一个parquet数据的备份。
❐ 对下机文件重新分析 ❐
如果说对于xenium的分割结果不满意,xenium ranger提供了重新分析的选项:
(1)调整分割参数,重新分割并生成结果文件(比如当有很多转录本在细胞外面时,可以选择增加expansion-distance,使得细胞更大,纳入更多的转录本);
(2)导入从QuPath、cellpose、baysor等重新分割后的mask掩码(格式为GeoJSON/tif/npy/json),生成对应的结果文件
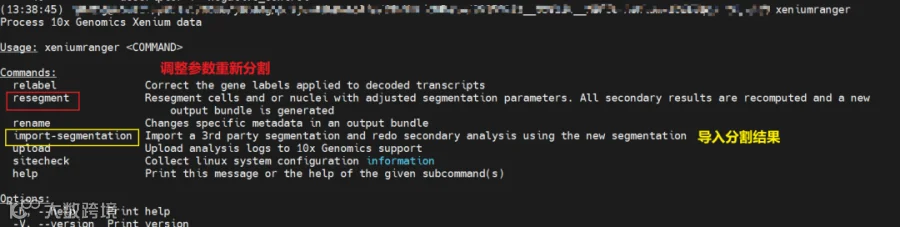
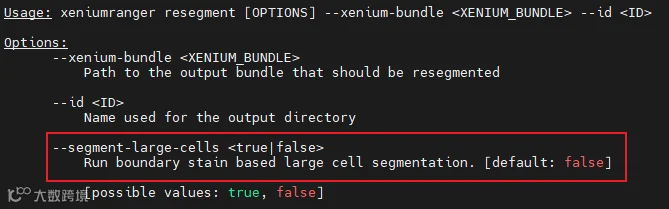
其中重新分割resegment在v4.0之后增加了分割大细胞的选项:
官方数据展示效果很不错(下图),如果大家的数据中有这种大细胞,可以尝试优化分割效果:
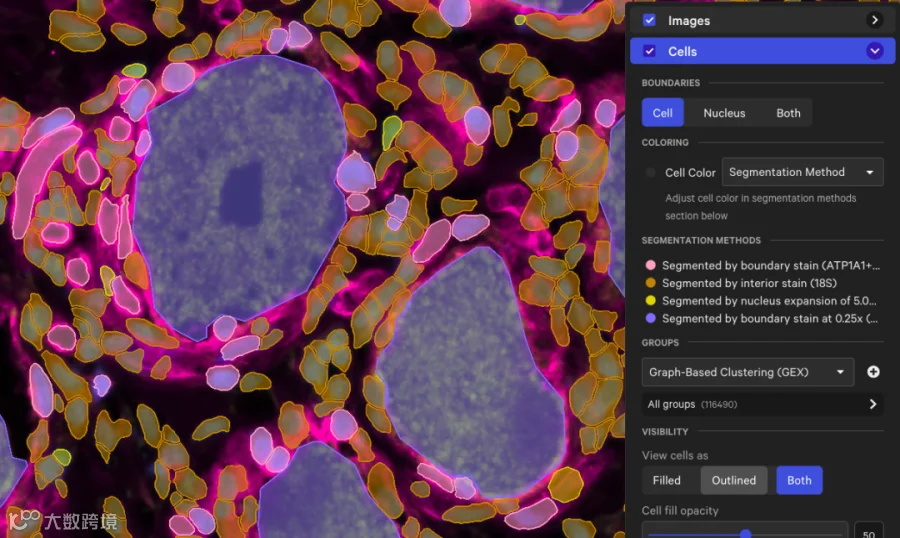
❐ 基因panel ❐
gene_panel.json这里面描述了panel中的所有基因、panel的信息,大多数基因都设计了2-3个探针。例如:
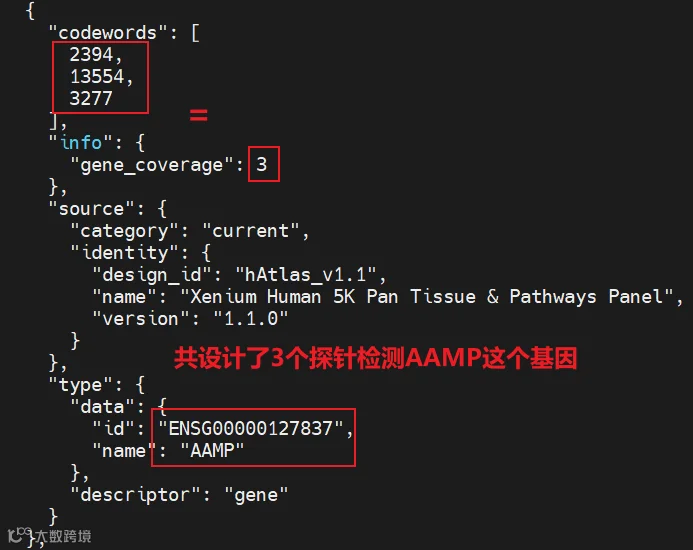
人的panel是5001个基因,小鼠的panel是5006个基因。两者的panel中的基因差别还是蛮大的。
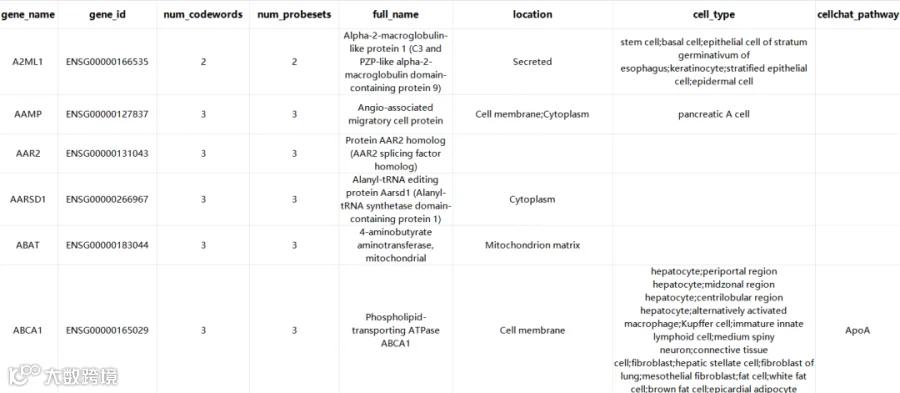
panel中基因的具体信息如下,其中一多半的基因标注了细胞类型,可以考虑作为细胞注释的参考:
❐ 结语 ❐
从本期开始,我们开启了xenium的空间转录组分析专题,初步内容包括:数据格式介绍、xenium explorer使用、xenium数据的分析、多样本整合、细胞注释、空间邻域分析、空间邻近度分析等,有丰富的代码和结果展示,带大家从入门到精通!
下一期介绍xenium explorer使用~
❐ 参考 ❐
【1】xenium下机数据官网详细说明:https://www.10xgenomics.com/cn/support/software/xenium-onboard-analysis/latest/analysis/xoa-output-understanding-outputs
【2】xenium 5kg enepanel的基因说明:
https://www.10xgenomics.com/support/software/xenium-panel-designer/latest/tutorials/pre-designed-xenium-prime-5k
【3】seurat官网中xenium数据的分析教程:
https://satijalab.org/seurat/articles/seurat5_spatial_vignette_2#mouse-brain-10x-genomics-xenium-in-situ
【4】squidpy官网中xenium数据的分析教程:
https://squidpy.readthedocs.io/en/stable/notebooks/tutorials/tutorial_xenium.html
👉 关于亿科羲和 👈
南京亿科羲和基因数据科技有限公司(XHGeneData),成立于2023年,是一家以数据科学与人工智能为核心驱动力的前沿生物科技企业。公司深度融合生物信息学与软件工程的跨学科技术,专注为生命科学研究和医疗健康领域提供创新的组学数据分析解决方案,致力推动精准医疗和生物医药研发的智能化进程。我们以“解码生命数据,赋能健康未来”为使命,通过技术革新持续探索生命数据的价值边界,引领行业智能化变革。
官网链接:https://www.xhgenedata.cn

