


第一作者:苏晓然,胡博韬,张英政
通讯作者:赵娣,张加涛,陈晨
通讯单位:北京理工大学,清华大学
论文DOI:10.1002/anie.202511671

利用可再生电力将CO2转化为CO,为合成各种化学品和燃料提供了一条可持续的途径。然而,由于缺乏普适性的电位适应性,大规模转化受到很大限制。基于此,北京理工大学赵娣特别研究员、张加涛教授和清华大学陈晨教授合作,成功开发了一种以富氧空位的ZnO为载体锚定CoPc的电催化剂(CoPc@ZnOv),从而提高了CO2到CO转化的活性和选择性。值得注意的是,CoPc@ZnOv在H池中于1.3 V的超宽电位窗口内CO法拉第效率保持在90%以上,在流动电池中为1.40 V,在MEA为1.0 V。其性能超过了先前报道的基于分子CoPc的电催化剂,甚至超过了大多数单金属位点材料。密度泛函理论计算结合原位光谱分析表明,由p-n结整流效应产生的内置轴向电场能够驱动具有不对称电荷分布和几何曲率的富电子单分子Co-N4位点,促进*COOH的形成、*CO的脱附,并抑制析氢反应,从而在超宽电位窗口内有利于通过CO2还原反应生成CO。这项工作提出了一种基于内置轴向电场理论的不对称单分子Co-N4位点的新型催化剂设计策略,同时调节面外极化以提高催化性能。

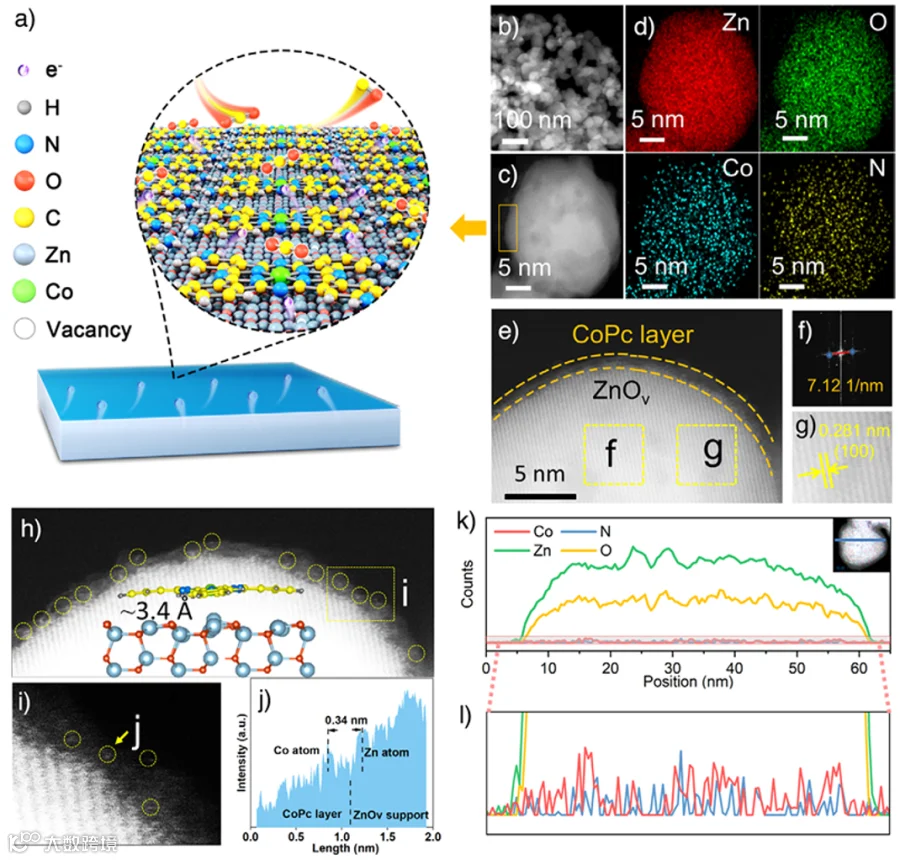
图1. 形态结构表征
首先通过煅烧的方式合成了具有丰富氧空位的ZnOv纳米粒子,然后使用简单的湿化学方法用CoPc对其进行修饰(图 1a)。高分辨率透射电子显微镜图像显示,CoPc@ZnOv和ZnOv具有相同的纳米粒子形态(图 1b)。采用高角度环形暗场扫描透射电子显微镜来分析CoPc@ZnOv的结构(图 1c)。EDS图谱显示,Zn、O、Co和N的元素均匀分布(图 1d)。放大图像清晰地显示CoPc层锚定在ZnOv外层(图 1e)。如图1f、g 所示,间距为0.281纳米的晶格条纹归因于ZnOv的(100)晶面。图1h、i展示了多个孤立的亮点,表明Co原子在CoPc@ZnOv表层中均匀分散。沿着黄色箭头所指的方向的强度表明,钴原子是单独存在的,并且与ZnOv层相距约3.4 Å(图 1j),这表明单原子钴与ZnO的边缘紧密接触。图1k和l进一步证实,CoPc是锚定在ZnOv外层上的。
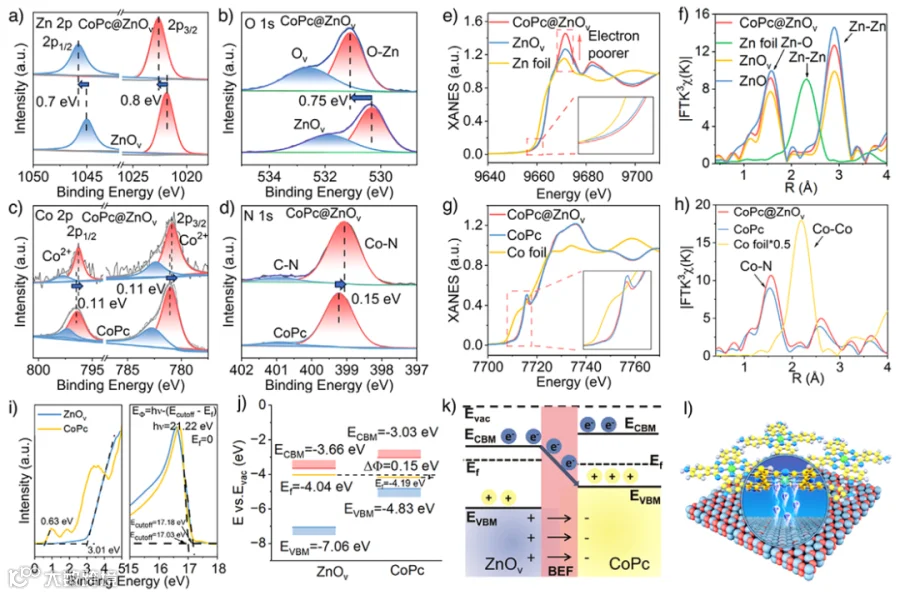
图2. 化学结构表征。
通过XPS进一步研究了CoPc@ZnOv的化学组成和价态。与ZnOv相比,CoPc@ZnOv中的Zn和O峰向更低的结合能方向移动(图2a-b)。相反,Co和N的峰位置与CoPc相比向更低的结合能方向移动(图2c-d),表明Co和N原子的电子富集。随后进行了XANES和EXAFS光谱分析。CoPc@ZnOv中锌的近边向更高的氧化态移动,这证实Zn原子周围电子密度的降低(图2e)。ZnOv表现出低于ZnO的锌氧键配位数,这进一步证明了氧空位的存在(图2f)。CoPc@ZnOv中的Co近边位置相对于CoPc左移,这可能是由于从ZnOv向CoPc的电子转移所致(图2g)。CoPc@ZnOv几乎没有在2.12 Å处的Co–Co键合,表明Co单原子分散性良好(图2h)。综合上述证据,证实了锌、氧、钴和氮原子之间电子密度的重新分布。
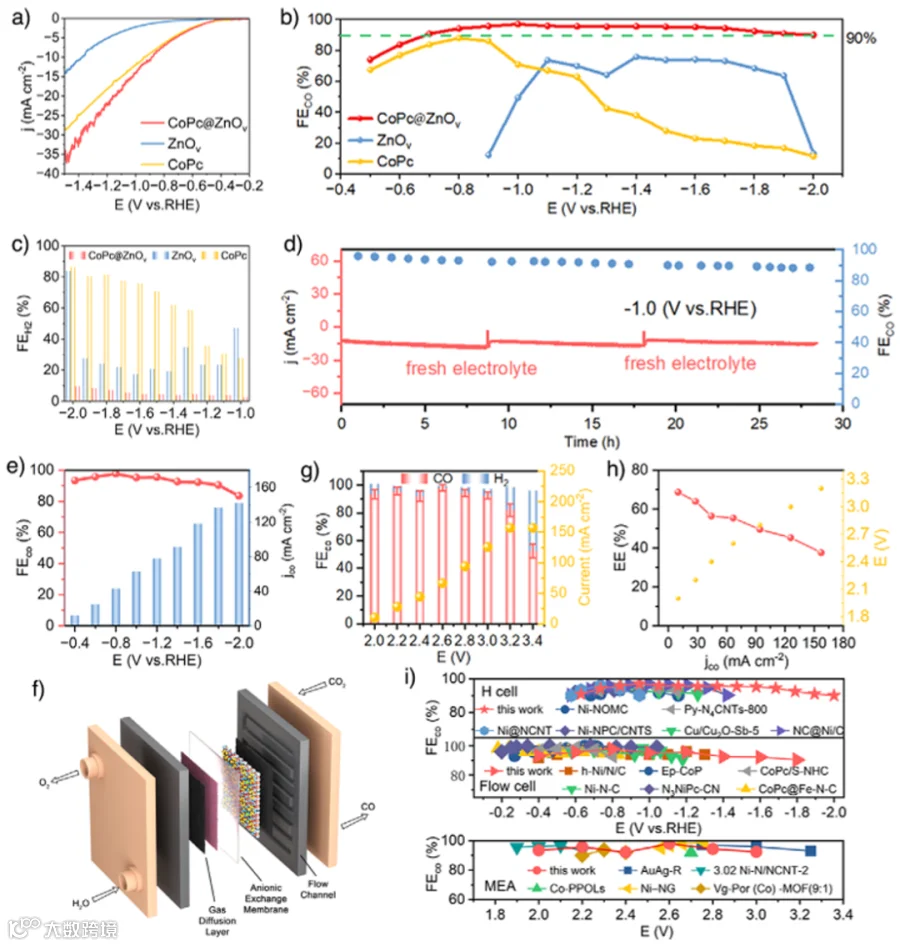
图3. CO2RR性能。
为了进一步探究CO2RR的性能,采用H池进行了测试。图3a表明CoPc@ZnOv具有最高的CO2RR活性。随后,在CoPc@ZnOv、ZnOv和CoPc上进行了一组恒电位电解实验。如图3b、c所示,CoPc@ZnOv的最大CO法拉第效率(FEco)可达到97.2%。更重要的是,在-0.7至-2.0 V的超宽电位窗口内,CO法拉第效率始终保持在90%以上。值得注意的是,FEco > 90%的电位窗口比先前报道的电催化剂的电位窗口更宽(图3i)。CoPc@ZnOv的电流密度在长达约30小时的长时间测试中保持了相对稳定的状态,而FEco也一直保持在90%以上(图3d)。流动池测试中,CoPc@ZnOv的FEco在超宽电位范围(-0.4至-1.8 V)内仍保持在90%以上(图3e)。基于GDE的零间隙MEA电解槽有利于在实际水平上提高CoPc@ZnOv的电流密度和法拉第效率(图3f)。在图3g中,最大FEco达到98.1%。在电压为2.0至3.0 V的范围内,FEco的值始终保持在90%以上。如图3h所示,CoPc@ZnOv在j分别为27.44、44.22、66.66和94.33 mA cm−2时,具有63.95%、56.43%、55.42%和49.52%的高能量效率。
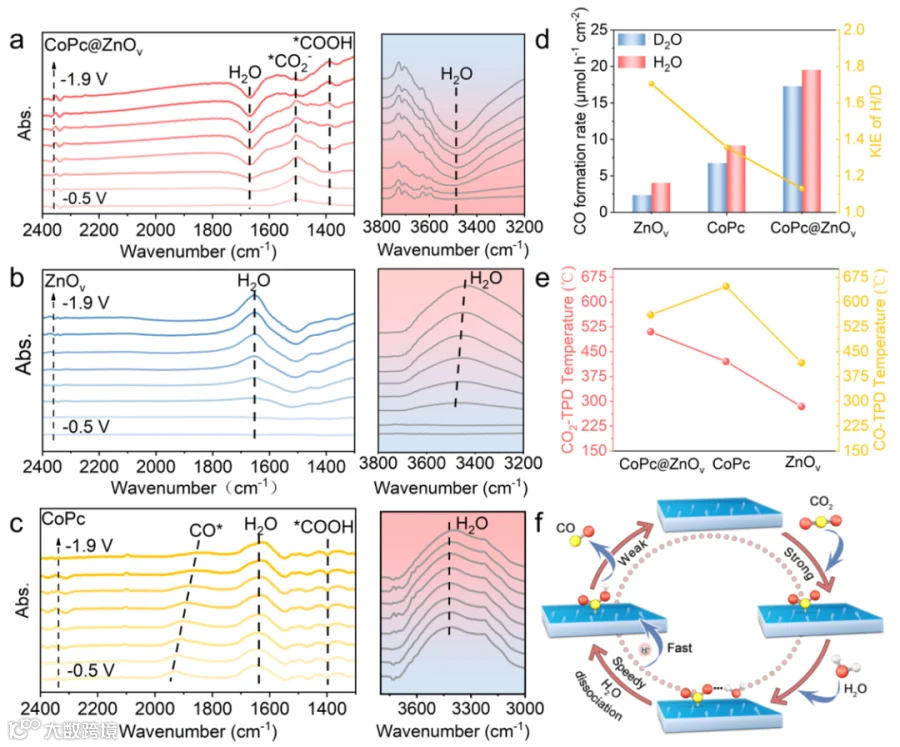
图4. 反应机理分析。
为了更好地理解CoPc@ZnOv上的CO2RR机制,使用原位ATR-SEIRAS来检测CO2RR过程。如图4a所示,在CoPc@ZnO中观察到1410 cm−1处的正峰和1620 cm−1处的负峰(分别对应吸附的 *COOH 和 H2O)在-0.5 V时出现。当施加的电位增加时,水分子首先被激活并持续被消耗,以提供质子来快速进行质子化,从而使*CO2−中间体变形,形成*COOH中间体,这通常是CO2RR在CO生成过程中的速率决定步骤。ZnOv表面约3300 cm−1处的水吸附峰值随着电位的降低而逐渐变为正值,这表明ZnOv表面的水持续富集(图 4b)。所以,形成质子是相当困难的。水分子的特征峰对于CoPc也是呈正向的,而且在1900 cm-1处检测到了吸附过程中*CO特征峰,这表明*CO的解吸较为困难,不利于CO的生成(图 4c)。为了进一步分析在CO2RR过程中的H2O分解过程中质子传输速率,进行了动力学同位素效应(KIE)分析。如图4d所示,ZnOv和CoPc的KIE值分别为1.7和1.38。对于CoPc@ZnOv,KIE值降至1.08,表明CoPc@ZnOv确实加速了质子转移过程,并进而增强了CO2RR的活性。图4e展示了CoPc@ZnOv、CoPc和ZnOv的化学CO2脱附峰分别位于520、412和275°C,这表明CO2在CoPc@ZnOv上的结合强度得到了增强。因此,提出了CoPc@ZnOv的反应机制(图 4f)。
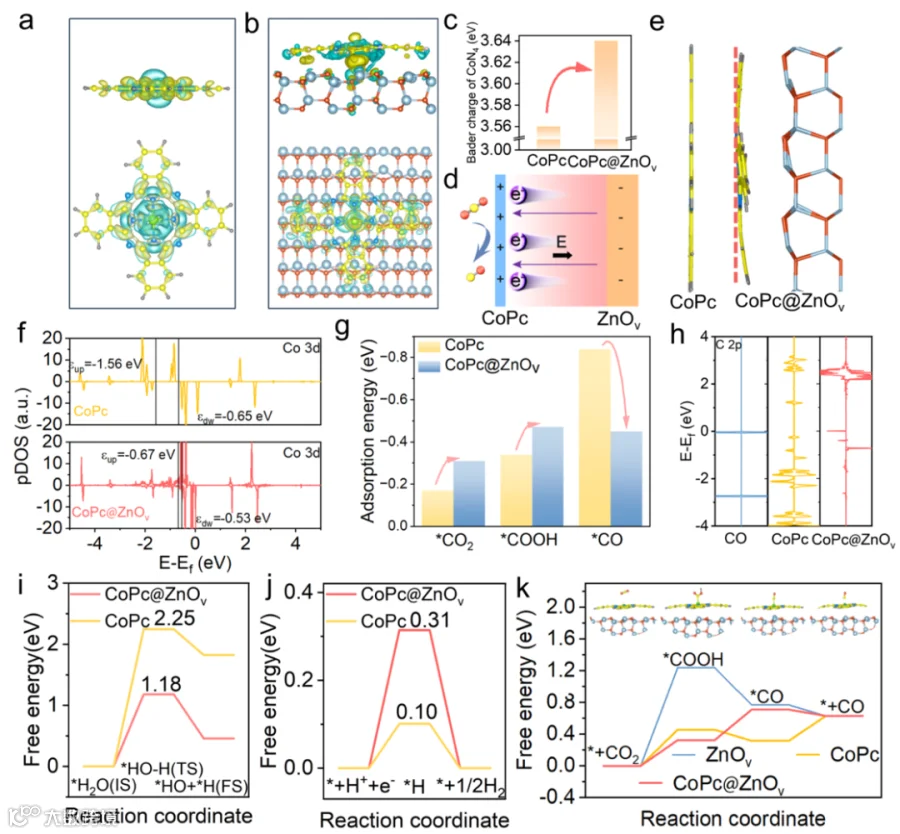
图5. 电化学活性位点及机制的阐明。
为了进一步阐明BEF在异质界面处对CoPc结构的影响,进行了DFT计算。具有特征平面Co-N4结构的CoPc表现出对称的电子结构。在CoPc@ZnOv中,电荷密度沿着CoPc和ZnOv之间的轴聚集。CoPc@ZnOv中的Co-N4中心通过轴向异质界面BEF效应从ZnOv表面获得了更多的电子,从而形成了一个更具电子丰富的Co-N4中心。轴向异质界面BEF效应导致的CoPc的弯曲是由于CoPc分子与ZnOv表面之间的范德华相互作用以及轴向平衡距离约为3.4 Å而形成的(图5e)。此外,轴向异质界面BEF效应导致Co 3d pDOS的不对称变化以及Co d轨道中心的上移(图5f)。如图5g所示在ZnOv表面的CoPc对*CO2和*COOH具有更强的吸附作用。图5h显示在CO-CoPc上观察到了更宽的C 2p峰,这表明表面吸附的物质具有更高的共价性,且纯CoPc的吸附作用更强,与上述吸附能结果一致。如图5i所示,CoPc@ZnOv的自由能为1.18 eV,低于纯CoPc的2.25 eV,这表明其H2O分解动力学显著加快,并具有出色的质子供应能力,有助于形成关键的COOH中间体。同时,CoPc@ZnOv的|△GH*|值(0.31 eV)表明其具有良好的反应动力学性能和出色的质子供应能力,有利于形成关键的COOH中间体。如图5k所示,ZnOv上Co-N4位点的RDS的自由能变化为0.39 eV(*COOH到*CO),这显著低于纯CoPc上Co-N4位点的自由能变化(0.46 eV,CO2到*COOH),因此ZnOv上的Co-N4位点在从热力学角度考虑时,对CO2到CO的转化具有出色的催化活性。

本研究成功地在p型单分子酞菁钴与n型富氧空位氧化锌间构建了异质结,从而提高了电催化CO2RR到CO的活性和选择性。CoPc@ZnOv的CO转化率在H池的超宽电位窗口、流电池的1.40 V以及MEA的1.0 V范围内都能保持在90%以上,超过了之前报道的分子催化剂的性能。机理表明,强烈的BEF轴向作用促使电子从ZnOv向具有富电子单个Co-N4位点的CoPc进行转移,这导致了单分子CoPc的不对称电荷分布和几何弯曲,从而调节了Co活性位点的d轨道中心。因此,这种轴向调节效应既增强了*COOH的吸附作用,又削弱了*CO的解吸作用,从而打破了它们之间的线性关系。此外,这种轴向的BEF效应对CoPc也促进了质子供应,通过H2O分解实现,并抑制了HER,从而有利于在超宽电位范围内选择性地通过CO2RR生成CO。这项研究为基于BEF理论在能源催化和存储设备中同时优化电子结构和几何结构提供了可能性。

Built-in Axial Electric Field-Driven Electron-Rich Monomolecular Co Sites for Promoting CO2 Electroreduction to CO Over Ultrawide Potential Window
https://doi.org/10.1002/anie.202511671
声明

