
第一作者:蒋琳
通讯作者:焦研教授
通讯单位:澳大利亚阿德莱德大学
论文DOI:10.1002/aenm.202303809
由于存在相互竞争的氢析出反应(HER),实现高活性和高选择性对电化学 N2还原是一项重大挑战。尽管基于密度泛函理论的计算可以识别出对eNRR有利的电催化剂,但理论预测与实验观察之间仍存在很大差距。本研究从动力学N2吸附、*N2加氢和相应的电位依赖动力学三个角度全面分析了eNRR和HER在电极-电解质界面上的动力学竞争。这些数据揭示了N2在EEI的吸附在动力学上是非常容易的。在*N2吸附后,随后的氢化受电极电位的影响。在较低的过电位下,*N2加氢比HER加氢更容易。然而,在较高的过电位下,由于 EEI 的N2供应有限,eNRR在动力学上处于劣势,而 HER 动力学则会加速并最终占主导地位。因此,确定了不同催化剂的电化学eNRR电位窗口。令人信服的证据表明,提高 EEI 附近的 N2 浓度是提高eNRR活性的关键。这些发现为旨在提高绿色氨合成效率的未来战略提供了重要的基本见解。
水电解质中的电化学还原氮气作为氨合成过程中传统哈伯-博世工艺的一种可行替代途径,前景十分广阔。然而,迄今为止观察到的氮气还原相对较低的选择性和活性仍是一个重大挑战,阻碍了其实际工业规模的生产。推进电化学氮气还原的主要障碍是同时在阴极侧发生的竞争副反应,即氢析出反应,电解质溶液中丰富的质子消耗电子并还原成副产物-氢气,从而从根本上破坏了eNRR的实用性能。因此,仅依靠热力学设计原理不足以有效解决HER带来的竞争问题。当务之急是了解eNRR和HER之间错综复杂的界面动力学,同时考虑到动力学参数。这种理解对于加快 eNRR 的开发至关重要,尤其是在实现更高的NH3生产选择性和活性方面。这种研究不应局限于考虑电催化剂的内在特性,还应包括应用电极电位以及电化学界面 (EEI) 上N2和质子的局部可利用性等因素。这些新发现的见解有望培育出超越以材料为中心的传统战略的替代设计原则,从而为 eNRR 的工业化实施铺平道路。
为了同时促进eNRR的活性与选择性,本文从动力学的角度出发,考虑了在电极-电解质界面上NRR的三个不同反应步骤与HER的相互竞争。
1. eNRR 活性差并不是氮吸附、活化或氢化反应本身较弱的结果,相反,氮吸附过程通常在动力学上很容易进行,而且氮可以在许多金属表面被活化,以进行随后的氢化反应。这种能力不受电催化剂内在电子结构特性的限制。
2. 由于H2 对电势高度敏感,因此在特定电势下,H2形成的能量势垒逐渐降低,由此导致HER的动力学加速并最终占主导地位。
eNRR 的第一步是二氮吸附到电催化剂表面的动态过程。这一过程包括将溶解的 N2 从电解质直接输送到催化剂表面,使电子转移到 N2 分子,从而削弱二氮三键。本文通过分析两种不同吸附构型(端面*NN和侧面*N=N*)的能量曲线 (ΔE),研究了 N2在各种过渡金属 (TM) 上吸附过程的动力学可行性,包括(110) 面的钯 (Pd)、铂 (Pt)、镍 (Ni)、铑 (Rh)、钌 (Ru) 和金 (Au)。如图 1a 所示,本次工作的计算模拟中,N2 分子最初被放置在距离催化剂表面 ≈ 4-5 Å 处,即所谓的亥姆霍兹层区域之外,直到 N2 被吸附在催化剂表面形成 TM-N 键。对于两种吸附构型,模拟结果表明,对于所有考察过的过渡金属,这种 N2吸附途径都是有利的,如图 1b & 1c 所示的能量曲线所示。当 N2 从 ≈5.0 Å 开始接近催化剂表面时,系统的总能量保持相对不变,直到距离达到 ≈3.0 Å,然后急剧下降。因此,在电化学还原过程中,这两种 N2 吸附途径对大多数过渡金属都是有利的。并且在Ru、Rh 和Ni这三种过渡金属中展示了较强的吸附能(图1d)。
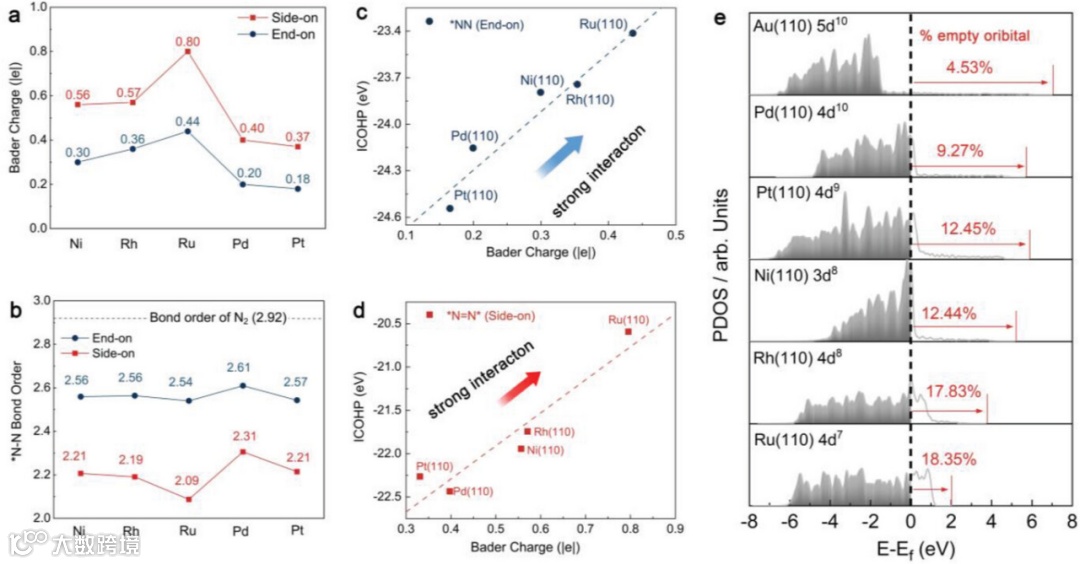
在研究了N2的动态吸附过程后,本文进一步对N2三键的活化程度在不同金属中进行了深入研究。为了解释 N2的强活化性、不同 TM 表面与吸附 N2活化强度之间的内在联系,本文探索了各种特性,如巴德电荷转移(图 2a)、*N2 的键序(图 2b)、集成晶体轨道汉密尔顿群(ICOHP)和 TM 表面的电子结构状态密度(DOS,图 2e)。本文证明了N2在 TM 表面的活化相对容易。分子轨道理论和 "反向捐赠 "理论可以解释 N2 活化与 TM 表面之间的基本关系。该理论认为,N≡N 三键通过吸附将电子密度从键本身捐献给未填充的 TM d 轨道,从而削弱了 N≡N 三键。比如,更接近费米级(0 eV)的 d 带中心与更低的 *N2 吸附能相关联,这表明吸附物与催化剂表面之间的相互作用更强。在所研究的过渡金属中,Ni、Rh 和 Ru反映了它们与 N2 和金属表面之间的强烈相互作用。

图3: NRR-HER氢化示意图

在本节中,本文重点讨论吸附二氮的所有可能氢化路径(图3),质子来源分别是溶解 H+和表面吸附*H,计算出了所研究的所有过渡金属氢化机制的自由动力学参数(ΔGa)和热力学参数(ΔG)(图4a-d)。并且采用了布伦斯特-埃文斯-波兰尼 (BEP) 关系将 ΔGa 和 ΔG 联系起来, 以全面探讨eNRR和HER的竞争力。研究表明,溶解的 H+ 是 eNRR 的主要质子源,这使得该过程在动力学上比 HER 更为可行。在所研究的金属中,Ru 是最活跃的催化剂,表现出最低的整体能垒。然而尽管具有可行性,目前 eNRR 的性能仍不能令人满意,需要进一步探索。由于外加电极电位在电化学催化反应中起着重要作用,因此研究电极电位对反应机理的影响对于提高 eNRR 的整体活性/选择性至关重要。

图5:NRR-HER电压依赖的第一步氢化的动力学竞争
上一节证明了 eNRR 在没有任何外加电位的情况下的可行动力学。本文进一步研究了涉及溶解 H+(图5a)和表面 *H(图5b)的每个基本反应的电极电位依赖行为,并将不同外加电位 U(相对于 RHE)下的活化能 ΔG‡ 作为关键指标。图5c 给出了每种催化剂(Ni、Rh 和 Ru)有利于 NH3生成的电化学电位窗口。并且,为了获得 HER 和 eNRR 之间与电位相关的动力学竞争,本文对三种关键电位下 *NNH 和 *H 形成的活化能和反应能变化进行了比较分析,包括参考基本反应电位 U0、0 V 与RHE 的关系,以及 Ni、Rh 和 Ru 的电极电位窗口下限(图5d-f)。在图5d 中,Ni 表面 *H 的形成在热力学上比 *NNH 的形成更有利;然而,两者的能垒相似,仅相差≈0.01 eV,这导致在所有电极电位下 HER 和 eNRR 之间都存在很强的竞争关系。与此相反(图5e),Rh 表面显示出与 Ni 相反的电催化性能:eNRR 中间体 *NNH 的形成比HER 中间体 *H 的形成更有利,两者的动能势垒有很大差异。这表明与 H2 演化相比,N2 还原过程在较低的现场电位以较快的速度进行。在 U = 0 V 条件下(相对于 RHE),*NNH 成为无势垒过程,而在相同条件下形成 *H 的势垒为 0.33 eV,这表明 *NNH 的形成更具竞争力。因此,正如之前的研究工作所证明的那样,Rh 是一种很有希望用于电催化 N2 还原反应的材料。如图5f 所示,在 U = 0 V 时,Ru 上 N2 还原过程的能量势垒为 0.09 eV(相对于 RHE)。尽管在这一特定电位下 *NNH 的形成仍然是非自发的(ΔG > 0 eV),但在高达 -0.65 V 的负电位范围内,eNRR 过程在动力学上比 HER 更有利。总之,本文发现了两种有利于 eNRR 的材料:Rh 和 Ru,它们具有相对较宽的电极电位窗口,并且在动力学上利于 *NN 加氢过程。另一个迹象是,当电极电位达到足够的负值时,所有三个研究步骤(eNRR的海洛夫斯基样步骤、HER 的沃尔默和海洛夫斯基步骤)的发生都没有障碍,这使得每个步骤的总速率取决于反应物浓度。

图6:NRR-HER的第一步加氢动力学分析

图7:微观动力学分析-反应物(质子和氮气)浓度对NRR的整体性能的影响
为了更好地理解反应速率与电位之间的关系(可通过实验测量),本文进一步使用过渡态理论(TST)计算了拟议氢化途径的电位速率常数曲线,如图6所示。此外,还利用微动力学建模(MKM)分析纳入了反应物浓度的影响,如图7所示。用性能最好的催化剂Ru作为评估,本文特别关注确定 eNRR 与 HER 竞争所需的 N2浓度水平。考虑到 N2 在水中的溶解度有限,在常温常压下,1 体积的水中仅溶解 0.02 体积的氮气,微观动力学建模利用电极表面 N2 与质子的对数比(以 log(CN2/C+)表示)以及电极电位。然后将这些因素与 *N2 和 *H 的界面覆盖率(θN2/θH,如图7a)以及稳态下的 eNRR FE 选择性(图7b)联系起来。结果表明,如图7a 所示,随着还原电极电位的增加,催化剂表面的 *H 覆盖率也会增加,并最终成为 HER 过程的主导。但是,如果界面溶解 N2 的浓度或可及性增加,则覆盖率会降低。这反过来又有利于 eNRR,因为 eNRR 中间体 *NxHy的覆盖率(x = 1, 2;y = 0, 1, 2, 3)会增加(图 S15,佐证资料),这是因为前一节研究的 N2 吸附和氢化过程非常容易,进一步抑制了 *H 的形成,从而导致 NH3 生产的高活性和高 NH3 选择性(图7b)。
鉴于这一事实,即使不受欢迎的副反应 HER 需要更高的活化能,丰富的氢源仍将使其在还原环境中占据主导地位,这一点至关重要。因此,在较高的负电位下,eNRR 很难与 HER 竞争,从而有利于 H2 的产生,并产生令人不满意的 NH3 选择性。本文从计算角度得出的结论得到了 Ru 基电催化剂上电化学 eNRR 实验结果的支持,实验结果列于 S4 部分(佐证资料)(表 S1,佐证资料)。可以看出,在相对较低的应用电位(< = -0.3 V 相对于 RHE)下可获得最大的选择性,而在较低的电位(尤其是-0.4 V 相对于 RHE)下可达到最大产率。还可以做出进一步规定,确保 *NN 而不是 *H 的高覆盖率,从而抑制通过 Tafel 机制形成 *HH。因此,提高 Ru 和 Rh 上 eNNR 整体活性的有效策略是确保反应物 *NN 的充足供应,即提高吸附 N2 的覆盖率。通过优化近电极区域的传质,在 eNRR 方面取得一定成功的研究也支持这一建议。例如,通过使用碱阳离子(Li+、K+、Na+ 和 Cs+)可提高电极-电解质界面的 N2 浓度;质子过滤共价有机框架(COF)的开发确保了通过过滤氮和活性质子源来提高活性和选择性。
本文中进行了大量的 DFT 计算,并分析了 eNRR 和 HER 在电极-电解质界面上的动力学竞争,考虑了氮的吸附/活化以及随后与 HER 的氢化竞争。结果证明,eNRR活性差并不是氮吸附、活化或氢化反应本身较弱的结果,相反,氮吸附过程通常在动力学上很容易进行,而且氮可以在许多金属表面被活化,以进行随后的氢化反应。这种能力不受电催化剂内在电子结构特性的限制。N2 还原的主要质子源是溶化氢 H+,其活化势垒通常低于 HER。然而,由于 H2对电势高度敏感,因此在特定电势下,H2 会成为主要质子源,并且没有能量势垒。根据这些发现,认为 eNRR 活性差的原因是水性电解质中氮的可用性低。正如微观动力学模型所示,反应物(N2)的浓度起着主导作用,而这方面的问题无法通过调节电压和改变催化剂表面的电子结构来解决。理想的 eNRR 反应条件应确保:1) 电催化剂对 N2 的强活化作用;以及 2) 在电极-电解质界面,尤其是在内层赫尔姆霍兹层中,N2浓度较高。进一步的开发工作应侧重于界面电解质,以提高 N2 浓度。本工作从原子层面揭示了 eNRR 的迟缓性质,并强调了优化 N2 可用性的潜在范围和策略,提供了可靠的计算证据,为 eNRR 技术的更多发展铺平了道路。
焦研,阿德莱德大学化学工程学院教授,澳大利亚研究理事会未来研究员。曾被评为澳大利亚40位科研新星之一,获澳大利亚政策与科学研究所颁发的杰出青年科学家奖。主要研究兴趣为计算电催化,即用包含密度泛函理论和分子动力学在内的多尺度模拟来研究不同催化剂表面发生的电催化反应,并以此为基础来设计更新的催化剂。这些催化剂主要应用于清洁能源转换领域,涉及到的反应有氧还原,氢析出,二氧化碳还原,氮还原和电池材料。已发表120余篇文章,共获超三万四千次引用,h因子为70。自2019年以来连续五年被科睿唯安评为化学方向的高被引学者。
课题组招收有计算背景的同学,有全奖支持(每年32,500澳元)。课题组注重创新、合作、和动手能力。有意者请将简历和英语成绩发送给yan.jiao@adelaide.edu.au。
可选的研究方向:
·使用计算设计电催化剂材料(二氧化碳还原,氮还原,碳氮偶联)
·使用动力学计算研究和设计电催化的局域环境
·机器学习设计新催化剂材料
欢迎关注我们,订阅更多最新消息 “邃瞳科学云”推出专业的自然科学直播服务啦!不仅直播团队专业,直播画面出色,而且传播渠道多,宣传效果佳。
“邃瞳科学云"平台正在收集、整理各类学术会议信息,欢迎学会、期刊、会议组织方择优在邃瞳平台上进行线上直播,希望藉此帮助广大科研人员跨越时空的限制,实现自由、畅通地交流互动。欢迎老师同学们提供会议信息(会有礼品赠送),学会、期刊、会议组织方商谈合作,均请联系王女士:18612651915(微信同)。
投稿、荐稿、爆料:Editor@scisight.cn
“邃瞳科学云”推出专业的自然科学直播服务啦!不仅直播团队专业,直播画面出色,而且传播渠道多,宣传效果佳。
“邃瞳科学云"平台正在收集、整理各类学术会议信息,欢迎学会、期刊、会议组织方择优在邃瞳平台上进行线上直播,希望藉此帮助广大科研人员跨越时空的限制,实现自由、畅通地交流互动。欢迎老师同学们提供会议信息(会有礼品赠送),学会、期刊、会议组织方商谈合作,均请联系王女士:18612651915(微信同)。
投稿、荐稿、爆料:Editor@scisight.cn















