
第一作者:张娟
通讯作者:李亚飞教授,王彧教授
通讯单位:南京师范大学
论文DOI:10.1021/jacs.4c05669
Cu单原子(Cu1)催化CO2/CO还原(CO(2)RR)为多碳产物(C2+)的构效关系仍然有许多争议。本工作通过密度泛函理论计算对Cu单原子催化剂(Cu SACs)的反应机理和机制进行了系统研究,揭示了Cu1原子无论在何种衬底上均不具备C2+产物活性。以稳定锚定在氮化碳上的Cu1位点(Cu1@C3N4)为例,由于缺乏邻近的活性位点,Cu1位点无法实现*CO的加氢以及CO−CO偶联反应,并且Cu1在反应条件下不稳定,容易浸出并聚集形成小的Cu团簇。动力学分析表明,当Cu1所衍生的团簇至少含有三个Cu原子时,才可以有效促进CO−CO偶联反应的发生。进一步将此模型催化剂扩展到其他典型的Cu SACs后,发现所有Cu1位点均无活性,而衍生的Cu簇的C2+性能则取决于衬底。本研究揭示了铜单原子催化CO(2)RR的内在活性起源,为未来催化剂的合理设计提供了理论指导。
通过电化学CO2/CO还原反应(CO(2)RR)生成高附加值的多碳化合物(C2+),不仅为储存间歇性可再生能源提供了新途径,而且在缓解化石资源枯竭方面展现出了广阔的应用前景。目前,铜基催化剂是已知的唯一在温和条件下表现出高CO(2)RR活性并能生成C2+产物的催化剂,但是在提高单一产物的选择性和转化效率方面仍面临诸多挑战。对于Cu基催化剂的调控,包括掺杂、形貌调控和局部配位环境优化等方面已有大量的研究。然而,不均一的活性位点和不清晰的构效关系阻碍了Cu基催化剂的合理优化。铜单原子催化剂(Cu SACs)具有明确的单原子Cu活性位点(Cu1),并呈现出产C2+产物的巨大潜力,因此近年来受到广泛的关注。然而,Cu SACs在实际条件下的构效关系仍然有许多争议。
1. 通过结合多种前沿的计算方法,阐明了Cu1@C3N4 SACs的Cu1位点由于缺乏邻近的吸附位点而不具备C2+活性,并且Cu1原子在反应过程中很容易浸出而聚集形成小团簇;
2. 计算揭示了对于Cu1衍生的团簇,需至少含有三个铜原子才能够有效促进CO−CO耦合反应,产生C2+活性;
3. 通过将模型拓展到其他不同衬底的Cu1催化剂上,证明其均不具备C2+活性,而衍生的铜簇催化剂的C2+催化性能则与衬底有关。
图1 (a) Cu1@C3N4的优化构型。(b) 30 ps AIMD模拟后的Cu1@C3N4与显式水模型的结构。(c) Cu1@C3N4上*CHO和*OCCO形成的自由能图。(d) Cu1@C3N4上形成*CHO中间体的能垒随电极电势的变化。
在Cu1@C3N4中Cu1原子与三个N原子结合形成CuN3结构(图1a),但在水溶液中Cu1原子会与一分子H2O吸附形成Cu1N2·H2O构型(图1b)。Cu1@C3N4上的Cu1位点无法有效促进*CO → *CHO,即使在恒电势的动力学分析中,其能垒依然很高;由于缺乏额外的活性位点,导致第二个CO分子无法吸附,使其同样难以进行CO−CO偶联反应(图1c-d)。因此,Cu1位点不是Cu1@C3N4 SACs生成C2+产物的真实位点。

图2 Cu1@C3N4中原始Cu1原子迁移至其相邻等效位置以及浸出的能垒。
通过“slow-growth”采样方法,作者评估了Cu1@C3N4中Cu1原子的结构稳定性。如图2a,b所示,Cu1原子可以轻易地从初始位置迁移到其相邻的等效位置,并且当其浸出0.90 Å时,原始的Cu-N键断裂,此过程需要约0.89 eV的能垒。上述计算表明Cu1原子在C3N4衬底中不稳定,容易浸出并聚集形成小的Cu团簇。

图3 Cux@C3N4 (x = 2−4)上*CHO以及*OCCO中间体形成的动力学分析。
图3的动力学分析表明Cux@C3N4 (x = 2−4)上CO−CO偶联的能垒低于形成*CHO中间体的能垒。在Cu2@C3N4上,由于第一个CO分子优先占据Cu2位点,CO偶联仍然在动力学上不利,而当Cu团簇中的原子数达到三个时,CO偶联变得动力学上可行,特别是Cu3@C3N4上的能垒仅为0.41 eV。因此,Cu1@C3N4SACs催化CO(2)RR生成C2+产物的真实活性位点至少需要三个Cu原子组成的团簇。
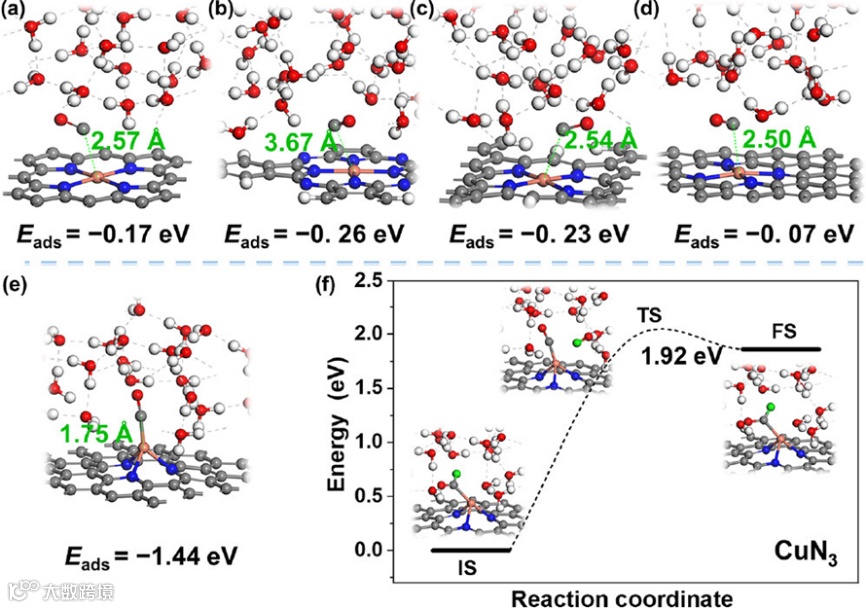
图4中作者将模型扩展到其他具有不同局部配位环境的Cu1中,对其衬底进行了研究。这些Cu SACs可以分为两类:(1)对于大多数实验中可获得的Cu SACs,它们的原始Cu1位点(CuN4结构)无法活化CO分子,表现出弱的物理相互作用;(2)尽管在某些催化剂的Cu1上CO分子可以被活化,但*CO → *CHO的能垒仍然很高,并且没有稳定吸附的*OCCO中间体形成。这些结果表明在不同衬底上的Cu1单原子催化剂同样不具备C2+产物的催化活性。而另一方面,衍生的Cu簇的C2+性能则受到衬底的影响(图5)。例如,Cu3@CuN3上CO−CO偶联的能垒高形成*CHO中间体的能垒,更利于产C1产物。

图5 Cu3@CuN3上*CHO以及*OCCO中间体形成的动力学分析。
本工作基于DFT计算、恒电势/杂化溶剂模型以及详细的动力学分析对Cu1@C3N4 SACs在电化学还原CO2/CO选择性生成C2+产物的机理上进行了深入的探究,揭示了由Cu1原子衍生出的、含有至少三个Cu原子的小团簇可能是真实的活性中心。与此同时,作者拓展的催化剂模型表明尽管Cu1原子在不同的衬底上均不具备C2+催化活性,但衍生Cu团簇的C2+性能与衬底相关。这项工作加深了对铜单原子催化剂上CO(2)RR机理的理解,阐明了Cu团簇作为真正活性中心对C2+产物选择性的关键影响,并为未来催化剂设计提供了指导。
张娟,南京师范大学李亚飞教授课题组博士研究生,研究方向为电化学CO2/CO还原模拟,在J. Am. Chem. Soc.、Energy Environ. Sci.等期刊发表论文4篇。
王彧,本科和博士毕业于南京师范大学(导师:李亚飞教授),毕业后在南洋理工大学从事博士后研究(合作导师:Zhou Kun教授),现为南京师范大学教授、博士生导师。研究方向为动态电催化模拟与设计,近五年以第一/通讯作者在在J. Am. Chem. Soc.、Nat. Commun.、Chem. Sci.等期刊发表论文40余篇。曾获得“江苏省优秀博士学位论文”(2019),入选江苏特聘教授(2022)和科睿唯安“全球高被引科学家”(2023)。
李亚飞,南京师范大学/常州大学教授、博士生导师。近年来致力于发展高效精准的电化学模拟方法来研究CO2电催化转化相关的机理与机制,并重点围绕“选择性转化机理”和“催化剂构筑策略”展开研究。近五年以通讯作者在Nat. Chem.、Nat. Catal.、Nat. Commun.、J. Am. Chem. Soc.、Angew. Chem. Ind. Ed.和Chem. Sci.等期刊上发表论文70余篇。所发表全部论文共被他引21000余次,个人h因子为81,2019年以来连续入选科睿唯安“全球高被引科学家”。担任Sci. Bull.和《科学通报》等学术期刊的编委。
欢迎关注我们,订阅更多最新消息 “邃瞳科学云”推出专业的自然科学直播服务啦!不仅直播团队专业,直播画面出色,而且传播渠道多,宣传效果佳。
“邃瞳科学云"平台正在收集、整理各类学术会议信息,欢迎学会、期刊、会议组织方择优在邃瞳平台上进行线上直播,希望藉此帮助广大科研人员跨越时空的限制,实现自由、畅通地交流互动。欢迎老师同学们提供会议信息(会有礼品赠送),学会、期刊、会议组织方商谈合作,均请联系潘经理:18612651915(微信同)。
投稿、荐稿、爆料:Editor@scisight.cn
“邃瞳科学云”推出专业的自然科学直播服务啦!不仅直播团队专业,直播画面出色,而且传播渠道多,宣传效果佳。
“邃瞳科学云"平台正在收集、整理各类学术会议信息,欢迎学会、期刊、会议组织方择优在邃瞳平台上进行线上直播,希望藉此帮助广大科研人员跨越时空的限制,实现自由、畅通地交流互动。欢迎老师同学们提供会议信息(会有礼品赠送),学会、期刊、会议组织方商谈合作,均请联系潘经理:18612651915(微信同)。
投稿、荐稿、爆料:Editor@scisight.cn













