第一作者:姚志波,程浩,许义飞
通讯作者:孙振宇,杨明,谭心怡
单位:北京化工大学,香港理工大学,北京理工大学
近日,来自北京化工大学的孙振宇教授,香港理工大学的杨明教授,以及北京理工大学的谭心怡教授合作,在国际知名期刊Nature Communications上发表题为“Hydrogen radical-boosted electrocatalytic CO2 reduction using Ni-partnered heteroatomic pairs”的研究文章。该研究构建了具有不同H吸附亲和性的Ni-配对杂双原子催化剂(HDACs),并将其应用于高效稳定的ECR。所开发的HDACs表现出非凡的催化活性和选择性,显著优于单原子电催化剂和许多先前报道的单原子电催化剂。基于理论和实验的综合研究,提出了氢自由基(H•)转移促进的化学加氢机理。这种尚未报道的途径与传统的第一PCET途径形成对比,后者需要高能量输入,从而显著提高了HDAC的ECR性能。研究还发现,通过选择金属(M)元素种类(即Cd、Pt和Pd)来调节M位点的H吸附强度,可以调节H•的形成,同时抑制析氢副反应,从而优化CO2还原速率。在~−0.75 V(相对于可逆氢电极,vs. RHE)下,CO的法拉第效率(FECO)高达97.1%,即使在连续55小时的CO2电解后也能保持良好的稳定性。在−200.0~−600.0 mA cm−2较宽电流密度(J)范围内,FECO超过90.0%。
由可再生电力驱动的电化学二氧化碳还原(ECR)有望将廉价和丰富的二氧化碳转化为增值化合物,同时也有助于实现可持续和低碳的未来。通过2e–转移过程产生的CO似乎是最有可能商业化的ECR产品。CO作为关键原料广泛应用于许多重要的工业过程中,包括水气变换反应、费托法和甲醇合成等。CO2到CO的电化学转化主要包含3个步骤: (1)CO2 + H+ + e−→ *COOH;(2)*COOH + H+ + e− → *CO + H2O;(3)*CO → CO + *(*表示吸附位点)。CO2到CO的转化通常受到第一质子耦合电子转移(PCET)形成*COOH的阻碍,这被认为是具有最高热力学势垒的电势决定步骤(PDS)。加速*H向CO2分子的生成和转移将是改善羰基中间体缓慢促进反应动力学的有效途径。为此,可构建具有双活性中心的电催化剂,其中H吸附和CO2加氢都发生在空间隔离的位置,但彼此位于原子尺度内。优化H和CO2在不同的双活性位点上的吸附,可加快*COOH形成的反应动力学,同时抑制竞争性析氢反应(HER),从而加快ECR。
要点1:利用原子捕获的方式,构建了具有不同H吸附能力的NiM-HDAC双原子催化剂,并将其应用于高效的电催化CO2还原。
要点2:基于理论和实验相结合的研究,我们提出了氢自由基(H•)转移促进的化学加氢机理。这种途径与传统的PCET途径形成对比,后者需要高能量输入,无法实现在低过电位下实现高的电流密度。我们还发现,通过选择元素(即Cd、Pt、Pd)来调节M位点的H吸附强度,可以调节H•的形成,优化CO2还原速率。
要点3:与可逆氢电极相比,在~−0.75 V下,CO的法拉第效率(FECO)高达97.1%,即使在连续55小时的CO2电解后也能保持良好的稳定性。过电位为566 mV时,实现优异的催化活性(JCO = −574.3 mA cm-2)。

图1:DFT计算研究NiM–HDAC催化剂的ECR反应机理。
密度泛函理论(DFT)计算首先使用NiM−HDAC (M = Cd、Pt、Pd)作为双原子催化模型。相对于Ni,所有的M位点都更容易将H2O解离成*H和OH–,并且H更容易吸附在M位点上。*H在M位点的吸附强度为:Cd > Pt > Pd。由图1a可知,E*COOH−E*H + CO2随着ΔG*H的增大而减小。即降低M位点的*H吸附强度(ΔG*H),可以降低Ni位点形成*COOH的能垒。这表明可以通过调节*H在M位点的吸附强度来调节ECR。以NiCdN6及其相关结构为模型催化剂,进一步研究了CO2转化为CO的反应。从计算得到的吉布斯自由能图(图1b)可以看出,对于所有催化剂来说,生成 *COOH都是PDS。如果假设Cd是CO2吸附和还原的活性位点,CdN4和NiCdN6(Cd位点)都具有最低的能垒。然而,计算出的HER的吉布斯自由能(图1c)显示,两种催化剂中的Cd位点更容易产生*H而不是*COOH。因此,我们提出带*H的NiCdN6代替NiCdN6(Cd位点)是最可能的反应模型。具体来说,Ni是ECR的活性中心,Cd是生成*H的主要位点。在确定反应模型后,通过计算CO2还原和H2生成的热力学极限势之差来估计催化选择性。基于这一指标,带*H的NiCdN6优于CdN4和NiCdN6(Cd位点)(图1d),表明其具有优越的ECR选择性。虽然与其他三种催化剂相比,NiN4对CO2还原具有更好的选择性,但它对*COOH的形成具有更高的屏障,从而阻碍了整体反应速率。同时,进一步进行催化剂与中间体电子结构的分析表明带*H的NiCdN6 对中间体有最优的吸附(图1e–i)。

通过原子捕获的方法,使用Ni–N结构捕获并稳定了Cd,形成NiCd−HDAC催化剂(图2a)。为了进一步揭示NiCd−HDAC的微观结构和分散性,采用高角环形暗场扫描透射电子显微镜(HAADF-STEM)进行了研究。高倍成像显示,许多单个原子形成原子对,测量到的原子对之间的平均距离为~0.27 nm(图2b,c),这意味着可能通过金属–金属(Ni–Cd)键和两个原子之间的强电子扰动形成异核原子结构。
采用同步辐射X射线吸收精细结构(XAFS)测量进一步研究了Ni和Cd在所得材料中的原子构型。在Ni K-edge FT-EXAFS光谱(图2d)中,NiCd−HDAC和Ni−SAC在1.87 Å处都有一个主峰,这是由Ni−N第一壳层配位引起。两种催化剂在2.48 Å处均不存在金属Ni−Ni键,进一步验证了金属的原子分散性。值得注意的是,在NiCd−HDAC光谱中可以观察到一条新的2.63 Å的散射路径,它既不是来自Ni−N(1.87 Å),也不是来自金属Ni−Ni(2.48 Å)。在Cd K-edge FT-EXAFS光谱(图2e)中,Cd−N第一壳层配位在2.27 Å处有一个优势峰。在Cd k边缘的NiCd-HDAC光谱中也发现了2.62 Å的路径,类似于在Ni k-边缘观察到的路径,这条路径可以合理地解释为Ni−Cd相互作用的贡献。

NiCd−HDAC在~−0.50 V到−0.99 V(相对于RHE)的整个电位窗口中,对CO的FE优于Ni−SAC和Cd−SAC,在−0.75 V(相对于RHE)时达到了~97.1±0.2%的FECO最大值(图3a)。基于电化学活性表面积(ECSA)对CO生成的部分几何电流密度进行了归一化。值得注意的是,在~ –0.50 V到–0.99 V的电位范围内,NiCd−HDAC具有明显高于Ni− SAC和Cd−SAC的ECSA标准化JCO值(相对于RHE),表明其具有更强的本征活性(图3b)。电化学阻抗谱(EIS)Nyquist图(图3c)表明,NiCd−HDAC具有明显低于Ni− SAC和Cd−SAC的界面电荷转移电阻(RCT),从而在ECR过程中提供最高的电子转移效率。此外,在电位窗口内,NiCd–HDAC的转换频率(TOF)值远高于Ni−SAC和Cd−SAC(图3d)。除此之外,NiCd−HDAC表现出良好的催化稳定性,即使在连续电解55小时后,电流衰减最小,FECO几乎不变(图3e)。为了改善H型电解反应器中CO2传质的限制,进一步采用一种带有气体扩散电极(GDE)的流动电池,以评估NiCd−HDAC在工业应用中的潜力。在1.0 M KHCO3和KOH溶液中,NiCd–HDAC在−200.0至−600.0 mA cm−2的宽电流密度范围内均获得了超过90.0%的FECO(图3f)。

为了深入了解催化剂表面的反应机理,通过控制添加0.1 M H2SO4溶液来调节阴极电解质的质子浓度。在pH≤3.3时,Cd−SAC不发生ECR反应(图4a)。在这些低pH值下NiCd–HDAC仍然保持90.0%的高CO的FE。我们发现Cd有助于生成H•,用5,5-二甲基-1-吡咯烷-N-氧化物(DMPO)作为自由基捕获试剂,通过原位电子顺磁共振(EPR)谱证实了这一点。在−0.6 V的氩气环境下,NiCd−HDAC清晰地观察到9个特征峰,对应的峰强度比为1:1:2:1:1:1:2:1:1(图4b)。相比之下,Ni−SAC没有获得EPR信号(图4b),这表明NiCd-HDAC中H•的形成归因于Cd。当引入CO2时,典型的9个信号消失,表明产生的H•在ECR过程中被中间体消耗。值得注意的是,NiCd-HDAC催化剂的FECO非常高,仅留下痕量的•COOH用于EPR检测。确实,出现了•COOH(AN = 17.4 G, AH = 14.4 G)的EPR信号(图4c),表明H•可以有效激活CO2生成•COOH。这些结果有力地证明了氢自由基在CO2还原中的关键作用。总的反应途径可归纳为:
(1) H2O + M + e−→ M−H + OH−
(2) M−H → M + H•
(3) H• + CO2 → •COOH
(4) •COOH + Ni → Ni−COOH
(5) Ni−COOH + H++ e− → Ni−CO + H2O

图5:NiCd-HDAC的电化学和操作性ATR-SEIRAS分析。
为了检查ECR过程中可能的反应中间体,进行了原位衰减全反射表面增强红外吸收光谱(ATR-SEIRAS)测量(图5a)。如图5b所示,在~1967 cm−1处的明显波段来自于线性吸附的*CO(*COL),当电位变化时(随着扫描电位从–1.1到0 V的变化,蓝移变化~26 cm−1),该波段出现斯塔克调谐现象。在~1788 cm−1处的峰值可归因于(*COOH)的C=O拉伸振动和/或桥接CO(*COB)的C≡O拉伸振动。然而,在图5b中,*COB和*COOH是否同时存在于该区域尚不明确。为了澄清这一点,我们进行了对照实验,在电解电位为–0.9 V(相对于RHE)的ATR-SEIRAS操作条件下,首先将CO2切换为CO,然后再切换回CO2(图6)。结果发现,当CO2切换为CO时,经过几次SEIRAS扫描后,由于*CO表面覆盖度的增加,*COL峰(~1975 cm−1)逐渐增强并发生蓝移,直到接近平衡;而低波数范围(从~1808到1814 cm−1)的峰强开始时随时间下降,然后稳定下来。这表明,较低的波数波段是来自于*COB和*COOH的共同贡献,峰强度的最初下降可能是由于*COOH物种的逐渐转化和解吸,使表面吸附的*COL浓度减少。当CO气氛切换回CO2环境时,高波数范围内的*COL峰变弱并发生红移,这可归因于*COL覆盖度的减少及相关的偶极子-偶极子相互作用的减弱。相反,低波数范围的峰值增加,表明CO2首先被还原为*COOH,然后在表面积累,在切换前平衡*COB覆盖度基础上而增强了峰强。尽管峰的位置随表面物质的组成和覆盖度而偏移而难以反褶积明确,但基于以上结果可推断出*COOH和*CO(*COL和*COB)中间体的形成。

图6:NiCd-HDAC的CO2-CO-CO2气氛切换ATR-SEIRAS分析。
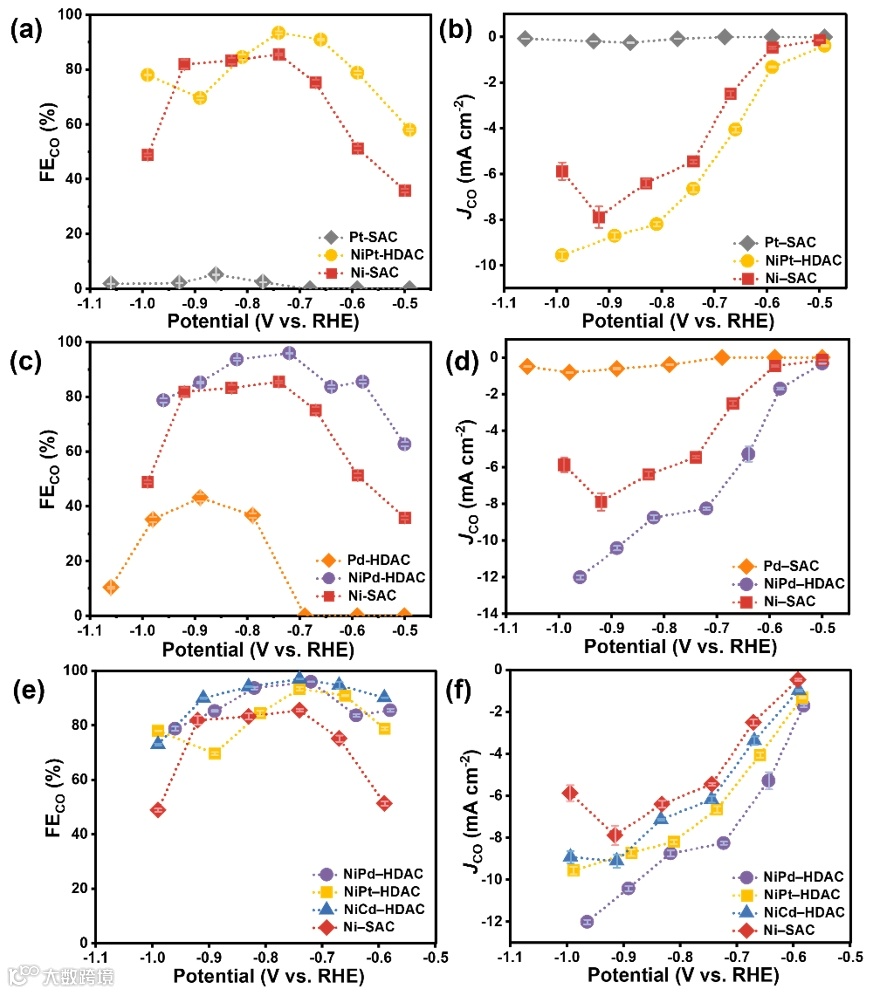
图7:不同NiM−HDAC(M = Cd、Pt、Pd)上的ECR性能。
与NiCd–HDAC类似,NiPt−HDAC和NiPd−HDAC在电压区域的FECO和JCO明显高于其相应的单原子对应物(图7),但NiPt−HDAC在~−0.89 V时的FECO比Ni−SAC低,这可能是由于NiPt−HDAC在该电位下的氢吸附最佳造成的,尽管与NiCd−HDAC相比,NiPt−HDAC和NiPd−HDAC上的HER倾向增加,但在整个电位范围内,两者均比NiCd−HDAC提供更高的JCO。
我们展示了合理设计的NiM−HDAC(M = Cd、Pt、Pd)增强ECR的H•转移途径。实验和DFT计算表明M是H•生成的活性位置,生成的H•进攻CO2形成•COOH,•COOH被Ni位点吸附,进行后续的电化学过程生成CO。通过选择合适的M位点和合适的H吸附强度,可以生成更多的H•,从而大大促进催化剂的反应动力学。我们提出的反应机理和催化剂设计概念为开发具有高活性和选择性CO2到CO转化的电催化剂提供了可能性。
Hydrogen radical-boosted electrocatalytic CO2 reduction using Ni-partnered heteroatomic pairs
https://www.nature.com/articles/s41467-024-53529-2
通讯作者:孙振宇,北京化工大学化工学院教授,博士生导师,新能源化工系主任。2006年博士毕业于中科院化学所。2006至2015年先后在爱尔兰圣三一学院、德国波鸿鲁尔大学以及英国牛津大学从事博士后研究。主要从事CO2、N2还原反应研究,以第一/通讯作者,在Nat. Common.、The Innov.、Chem、Angew. Chem.、Adv. Mater.、Prog. Mater. Sci.、Matter等期刊发表论文140余篇,英文书籍6章节,申请/授权发明专利17项。16篇一作/通讯作者论文入选ESI高被引论文,所有论文被Science等引用24000余次,H指数65。入选Elsevier2021–2023年度“中国高被引学者”,研究工作获选“细胞出版社2019 中国年度论文”、“2019 年中国百篇最具影响国际学术论文”。担任RSC Sustainability期刊副主编,The Innovation Materials 联合创刊人/学术编辑,《催化学报》、《物理化学学报》、ChemElectroChem等编委,中国化学会高压化学专委会委员,中国化学会二氧化碳化学专委会委员。瑞士、智利国家自然科学基金、德国洪堡基金等评委;Nature等期刊审稿人。
课题组介绍:https://www.x-mol.com/groups/ZhenyuSun
通讯作者:杨明,香港理工大学应用物理系助理教授、博士生导师,目前担任IEEE电子器件学会香港分会秘书。在Science、Nat. Phys.、Nat. Nanotech.、Nat. Common.、Phys. Rev. Lett.、Phys. Rev. B、Appl. Phys. Lett.、J. Am. Chem. Soc.等国际著名期刊上发表了超过125篇论文,引用超3600次,H因子约为36,并申请了3项专利。
通讯作者:谭心怡,长期从事二氧化碳电催化还原、电催化合成氨等电极材料以及锂离子、钠离子电池的高性能电极材料方面的研究和教学工作。承担国家自然科学基金青年基金、国家重点研发计划等多项项目。以第一作者/通讯作者身份发表学术论文20余篇,其中包括Nature Communications、Joule、Advanced Energy Materials、Advanced Functional Materials、Matter、Angewandte Chemie、Chemical Society Review等。
第一作者:姚志波,北京化工大学孙振宇教授课题组硕士研究生,现于上海交通大学李俊教授课题组攻读博士。研究方向为电催化二氧化碳还原研究。以第一作者身份在Nature Communications、Advanced Functional Materials等期刊发表4篇论文。
第一作者:程浩,香港理工大学应用物理系杨明助理教授课题组博士后,研究方向为新型多功能纳米催化材料的理论计算和机器学习应用。在Phys. Rev. Lett.、Nat. Mater.、Nat. Commun.、Energy Environ. Sci.、PNAS、ACS Catal.、ACS Appl. Mater. Interfaces、Angew. Chem. Int. Ed.等国际著名期刊上发表论文17余篇。
第一作者:许义飞,北京大学化学与分子工程学院在读博士生,研究方向为电化学固液界面性质及电极反应动力学,以第一作者身份在Nat. Catal.、Angew. Chem.、ACS Catal.等期刊发表6篇论文。
欢迎关注我们,订阅更多最新消息 “邃瞳科学云”推出专业的自然科学直播服务啦!不仅直播团队专业,直播画面出色,而且传播渠道多,宣传效果佳。
“邃瞳科学云"平台正在收集、整理各类学术会议信息,欢迎学会、期刊、会议组织方择优在邃瞳平台上进行线上直播,希望藉此帮助广大科研人员跨越时空的限制,实现自由、畅通地交流互动。欢迎老师同学们提供会议信息(会有礼品赠送),学会、期刊、会议组织方商谈合作,均请联系18612651915(微信同)。
投稿、荐稿、爆料:Editor@scisight.cn
“邃瞳科学云”推出专业的自然科学直播服务啦!不仅直播团队专业,直播画面出色,而且传播渠道多,宣传效果佳。
“邃瞳科学云"平台正在收集、整理各类学术会议信息,欢迎学会、期刊、会议组织方择优在邃瞳平台上进行线上直播,希望藉此帮助广大科研人员跨越时空的限制,实现自由、畅通地交流互动。欢迎老师同学们提供会议信息(会有礼品赠送),学会、期刊、会议组织方商谈合作,均请联系18612651915(微信同)。
投稿、荐稿、爆料:Editor@scisight.cn














