


第一作者:Hongling Yang、Xiaoxu Wang
通讯作者:陈晨、陈昕
通讯单位:清华大学、北京科技大学
DOI: 10.1021/jacs.2c11380

开发高效且简单的催化剂以揭示乙烯环氧化反应中的关键科学问题,一直是化学家所追求的长期目标;在该方面,设计非均相类分子催化剂是一种理想选择,因其结合了均相和非均相催化剂的双重优势。得益于明确定义的原子结构和配位环境,单原子催化剂可以有效模拟分子催化剂。在本文中,作者设计出一种由铱单原子组成的非均相催化剂,其可与反应物分子发生相互作用形成类分子催化特性,从而表现出优异的乙烯选择性环氧化反应性能,所获得高附加值环氧乙烷的选择性高达99%。研究表明,该铱单原子催化剂可有效提高环氧乙烷产物选择性的原因,可归因于具有较高氧化态的铱金属中心与乙烯或分子氧之间的π-配位。吸附在铱单原子位点上的分子氧不仅有助于加强乙烯分子的吸附,而且还可以改变其电子结构,使铱将电子注入乙烯的双键π*轨道。该催化策略有助于形成五元氧杂金属环中间体,导致环氧乙烷具有极高的选择性。本文设计出的单原子催化剂模型具有明显的类分子催化特性,可作为抑制所需产物过度氧化的有效策略。同时,将均相催化的概念应用于非均相催化,将为新型先进催化剂的设计提供一种新视角。

非均相催化剂因其易于回收和运行等优势而被广泛应用于工业催化领域,但其通常具有复杂的结构,使得难以精确设计和调控催化活性位点及其周围环境。相比之下,均相催化剂具有明确定义的配位几何结构、均匀的活性中心,可精准调控的配位环境等优势,但这类催化剂面临着可回收性差和产物分离难等问题。在过去的几十年里,科研人员设计出各种策略以将均相催化和非均相催化的优势相结合,即将均相催化剂的机理概念应用至非均相催化剂中。在这方面,迄今为止研究最多的策略是将分子金属络合物锚定在固态载体上,使表面附近形成典型的有机金属中心,从而赋予催化剂在各种催化转化过程中的可重复使用性。此外,纳米晶固定后催化剂的可回收性和产物分离性已取得较大进展,这些方法有助于理解均相催化和非均相催化之间的关系。然而,在连接上述两个催化领域方面,仍然有许多工作有待开展。就像在均相催化反应中采用非均相催化剂一样,在非均相转化中采用类均相催化剂不仅可以保持高活性和选择性,而且可以减少贵金属的用量。
从概念上讲,结合均相和非均相特征的催化剂由明确定义的活性位点组成,并且在反应过程中不溶于反应介质。近年来,单原子催化剂(SACs)为均相催化和非均相催化领域之间搭建了桥梁,得益于其明确定义的活性位点结构和易于进行的机理研究,被广泛应用于在分子水平上研究各种化学反应的结构-活性关系。与纳米颗粒催化剂相比,SACs通常表现出不同的化学性质和反应途径;这些差异是因为SACs中的金属位点通常携带部分正电荷,从而影响其电子密度、金属与反应组分之间的相互作用,甚至影响反应中间体的吸附行为。当SACs和反应物之间存在独特的配位结构时,可以预期催化系统将具有专门针对特定产物的选择性。
多相催化氧化是广泛应用于工业过程中的一类重要反应,例如乙烯选择性氧化为高附加值环氧乙烷(EO),尽管该反应已得到数十年的广泛研究,但通常以牺牲催化活性为代价实现高选择性。由于存在竞争性的过氧化副反应,同时实现高选择性和高活性仍然是一个巨大的挑战。银被认为该反应最为有效的催化剂,并被广泛研究。到目前为止,EO的选择性可达到90%,但该性能仅在促进剂(如Cs、Re、C2H4Cl2、Cu和Au)的帮助下才能实现,并且该反应的关键催化机制尚未达成共识。此外,对乙烯环氧化反应过程进行量子化学计算和原位光谱的研究非常稀少,这可能是优于缺乏既具有优异性能又具有良好结构的新型催化体系所致。
铱在均相有机反应如烯烃或炔烃转化中表现出独特的性能,铱盐或铱络合物([{Ir(OH)(cod)}2], [{IrCl(cod)}2], [{Ir(OMe)(cod)}2], [HIr(cod)(dppm)] (cod = 环辛二烯, dppm = Ph2PCH2PPh2))主要通过形成π-烯烃或π-炔烃组分来催化上述反应,从而选择性地获得所需产物。考虑到烯烃在金属活性中心上的化学吸附模型与均相金属-烯烃络合物的经典Dewar–Chat–Duncanson模型相似,将均相催化的概念应用至非均相催化体系中以解决乙烯环氧化反应的关键问题,有望实现仅依靠非均相催化无法达到的效果。

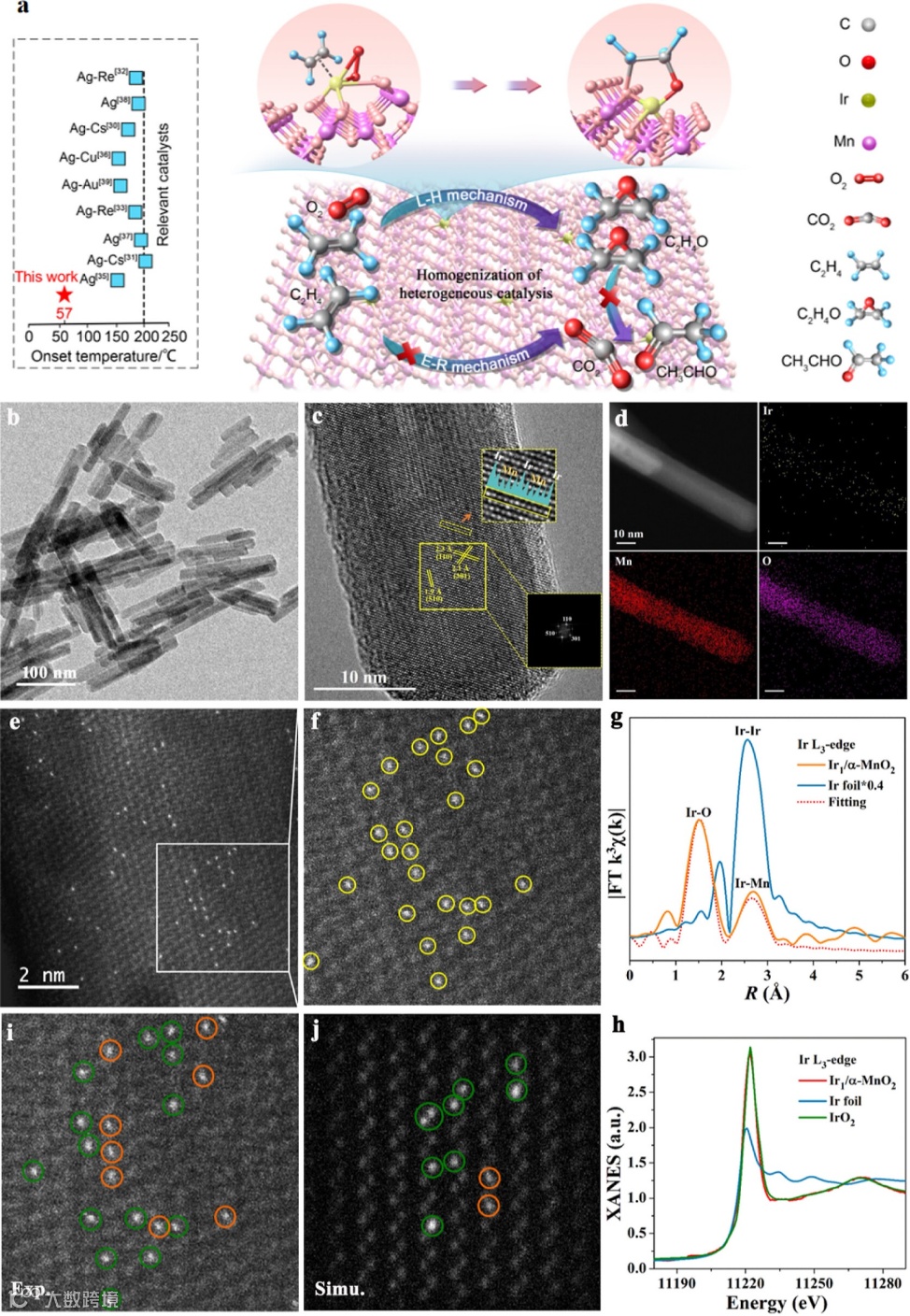
图1. (a) Ir1/α-MnO2催化乙烯环氧化反应的示意图。Ir1/α-MnO2的(b) TEM和(c) HRTEM图。(d) Ir1/α-MnO2的HRTEM图及相应的元素映射结果。(e,f) Ir1/α-MnO2的AC HAADF-STEM图。(g) Ir1/α-MnO2和Ir箔的Ir L3-edge FT-EXAFS谱。(h) Ir1/α-MnO2, Ir箔, IrO2的Ir L3-edge XANES谱。(i) Ir1/α-MnO2的高倍率HAADF-STEM图和(j)相应的模拟图(绿色圆圈表示取代铱位点,橙色圆圈表示吸附铱位点)。

图2. Ir1/α-MnO2, Ir NPs/α-MnO2, Ag1/α-MnO2, Ag NPs/α-Al2O3催化乙烯环氧化反应的(a)乙烯转化率,(b)产物分布,(c) Arrhenius曲线。AA和EO分别代表乙醛和环氧乙烷。(d)稳定性测试后Ir1/α-MnO2的AC HAADF-STEM图。(e)稳定性测试后Ir1/α-MnO2, Ir箔和IrO2的Ir L3-edge XANES谱。(f)稳定性测试前后Ir1/α-MnO2和Ir箔的FT-EXAFS谱。

图3. (a)在−173, −143, −103和−63 °C温度下,吸附质在Ir1/α-MnO2上的原位IR光谱。(b,c)吸附Ir1/α-MnO2的DOS计算。(d) Ir-O2-乙烯中碳和铱之间相互作用的COHP分析。(e) (i) C1–Ir–C2 π键, (ii) O1–Ir–O2 π键, (iii) O–O 2c–2e键的SSAdNDP化学键模式。(f)吸附α-MnO2、氧和乙烯之间的电荷密度差分图,其中黄色和青色区域分别表示电子积聚与耗尽。(g)在−173 °C的不同预处理条件下,于全饱和覆盖下CO吸附的原位IR光谱。(h)在250 °C的不同气体处理下,Ir1/α-MnO2的原位XPS谱。虚线为室温超高真空条件下Ir1/α-MnO2的Ir 4f峰位置。(i) Ir1/α-MnO2上18O2与乙烯反应的TPRS。

图4. (a) Ir1/α-MnO2在250 °C的乙烯/O2/He气氛下的原位IR光谱。(b)在乙烯环氧化反应过程中,Ir1/α-MnO2于不同气氛下的Ir L3-edge XANES谱。(c)在不同气氛下Ir1/α-MnO2, Ir箔和IrO2的EXAFS-FT谱。(d) Ir1/α-MnO2的白线峰位置与d带空穴数之间的关系。

图5. 在16O2/18O2/He气氛和不同温度下,(a) Ir1/α-MnO2和(c) Ag NPs/α-Al2O3的氧同位素测试。在16O2/18O2/He和乙烯/He的混合物气氛与不同温度下,(b) Ir1/α-MnO2和(d) Ag NPs/α-Al2O3的氧同位素测试。

图6. 根据(a)基于铱与乙烯或分子氧之间的π配位结构的机制和(b)传统的E–R机制,所计算出Ir1/α-MnO2催化剂上乙烯环氧化反应的自由能。

总的来说,本文开发出一种新型乙烯环氧化催化策略,即通过固定于α-MnO2纳米棒上的非均相铱单原子(Ir1/α-MnO2)与乙烯和分子氧相互作用,形成一种类分子催化剂,其催化特性接近分子催化,与常规的铱纳米颗粒催化剂不同。该Ir1/α-MnO2表现出优异的乙烯环氧化催化活性,为Ag NPs/α-Al2O3催化剂的7.6倍,EO产物选择性高达99%。原位测试和量子化学计算表明,Ir、乙烯和分子氧之间的π-配位结构可促进五元氧杂金属环中间体的形成,并加速生成EO产物。此外,该反应机制也不同于Ag基催化剂的传统E–R机制。该研究提供了对氧化物负载单原子在实际催化条件下局部结构的深入理解,同时这种精心设计的催化策略也可用于调节金属的氧化性能。此外,将均相催化的概念应用至气-固相催化反应中,将为新型先进催化剂的设计提供一种新视角。
【文献来源】
Hongling Yang, Xiaoxu Wang, Qinggang Liu, Aijian Huang, Xun Zhang, Yi Yu, Zewen Zhuang, Ganggang Li, Yang Li, Qing Peng, Xin Chen, Hai Xiao, and Chen Chen. Heterogeneous Iridium Single-Atom Molecular-like Catalysis for Epoxidation of Ethylene. J. Am. Chem. Soc. 2023. DOI: 10.1021/jacs.2c11380.
文献链接:https://doi.org/10.1021/jacs.2c11380
声明

“邃瞳科学云”直播服务

扫描二维码下载
邃瞳科学云APP



