
第一作者:赵文茹
通讯作者:梅东海教授,柳伟
通讯单位:天津工业大学材料科学与工程学院,天津工业大学环境科学与工程学院,中国石化大连石油石化研究院
论文DOI:10.1021/acscatal.4c01706
多环芳烃(PAHs)加氢裂化生产苯、甲苯和二甲苯(BTX)是最重要的工业石化过程之一。尽管经历了广泛的实验和工业实践,但多环芳烃在沸石负载的金属催化剂上的加氢裂化反应机理仍然难以捉摸。特别是PAHs的芳环如何通过C−C键断裂打开尚不清楚。在本工作中,以HY分子筛封装的单原子Pt催化剂(Pt1/HY)为例,利用密度泛函理论(DFT)计算研究了蒽在Pt1/HY上的加氢裂化反应路线。系统地研究了蒽的末端环和中心环的开环过程与芳环和氢源的饱和加氢程度之间的关系,从而导致不同的BTX产物。氢转移在同一芳环上是容易的,而在相邻芳环之间的连接C原子(Ca)处则受到动力学阻碍。在2,3-二氢蒽异构化为9,10-二氢蒽的情况下,末端和中心环之间的氢转移可以在蒽的帮助下通过分子间氢转移机制实现。与从Pt1位点加氢过程相比,在部分氢饱和的蒽芳环上添加来自Brønsted酸性位点(BAS)的质子会明显削弱C−C键,从而导致中心开环过程。此外,本工作还研究了以辛基蒽为探针分子的带支链多环芳烃的加氢裂化反应机理。结果表明,辛基蒽在环-支链连接位置的去烷基化在动力学上比中心开环和辛基链的裂解更可行。
催化加氢裂化工艺被广泛认为是一种高效的方法,通过在氢气存在下通过C−C键断裂减少环结构数量,将多环芳烃(PAHs)转化为苯、甲苯和二甲苯(BTX)等有价值化学品。在工业加氢裂化过程中,通常使用固体酸基双功能催化剂,特别是由大孔沸石负载的金属。沸石中的Brønsted酸性位点(BAS)有助于裂化和异构化,而活性金属组分在加氢、脱氢和异构化中起着至关重要的作用。然而,由于多环芳烃的复杂性质以及活性金属、酸性位点和促进剂的协同作用,在双功能酸性金属催化剂上全面研究多环芳烃加氢裂化的反应机理、本征动力学和结构-性能关系具有挑战性。
1. 采用密度泛函理论计算,确定了在实验条件下封装的Pt2+离子在HY沸石中的β笼六圆环(6MR)上的稳定性。在此基础上,系统地研究了多环芳香烃在Pt1/HY催化剂上的加氢裂化反应机理。
2. “C-a位”是阻碍了氢原子从末端环转移到中心环。2,3-二氢蒽异构化为9,10-二氢蒽是在附近蒽分子的辅助下通过分子间氢转移发生的。
3. 探讨了单原子金属Pt1催化剂和双功能催化剂(Pt1/HY)在多环芳烃加氢裂化反应中的差异,Pt1和Pt1/BAS位点上蒽加氢裂化产物不完全相同。此外,蒽的中间环开环反应速率高于末端开环反应速率。
4. 探究了支链多环芳烃(2-辛基蒽)的加氢裂化,证明了支链多环芳烃在末端环和支链烃链链接处的加氢裂化比多环芳烃的中心环开环更容易。
本研究首先对氢气在Pt1/HY位点上的吸附和活化进行了探讨。在典型的反应温度(773 K)下,我们研究了涉及不同共吸附反应中间体的四种氢吸附情况。结果显示,在裸露的Pt1位点上,氢吸附的计算吉布斯自由能为+42.9 kJ/mol。然而,对于共吸附的蒽和9,10-二氢蒽,由于Pt1位点和共吸附的中间体之间的强相互作用,氢吸附在热力学上变得更加不利。计算结果显示,共吸附反应中间体的存在会影响吸附的氢分子的解离,具体而言,在Pt1位点氢离解的活化吉布斯自由能从不存在共吸附中间体时的73.1 kJ/mol降低到存在共吸附的1-苄基-2-甲基苯时的36.2 kJ/mol。

图1 氢气在Pt1/HY沸石的Pt1位点吸附和离解的吉布斯自由能谱
根据研究结果,我们对第一个氢原子添加到吸附的蒽中的情况进行了深入分析,特别关注了中心环和末端环之间的潜在氢转移。实验结果显示,在中心环的C9位置加入第一个氢原子在热力学上是非常有利的,其能为−36.5 kJ/mol。相反,在末端环的C1位置添加第一个氢原子则产生了轻微的偏转角,吉布斯自由能变化为+3.3 kJ/mol。此外,我们计算出氢原子从C9转移到C9a的活化吉布斯自由能和反应能分别为142.0 kJ/mol和+23.8 kJ/mol,表明氢转移过程是吸热过程,并存在显著的动力学屏障。类似地,在末端环的C1位置添加第一个氢原子导致从C1到C9a的高吸热(+71.8 kJ/mol)的氢转移,伴随着150.1 kJ/mol的高活化势垒。同时,我们发现在蒽的同一末端环内,氢原子从C1位置转移到相邻的C2位置在热力学上是中性的,在动力学上也是可行的,活化势垒为45.3 kJ/mol。因此,我们推断蒽环内的氢转移过程更可能发生在同一环内,而不是相邻环之间。这些发现为进一步研究蒽分子的反应性和催化性能提供了重要参考。

图2 在最初氢化的蒽上的C9 → C9a、C1 → C9a和 C1 → C2位置之间的氢转移过程
随后对2,3-二氢蒽进行了氢转移过程异构化为9,10-二氢蒽的研究。实验结果显示,在同一末端环内,氢化物从C3位置转移到C4位置的活化势垒相对较低,为27.1 kJ/mol,且该转移步骤释放能量为-49.7 kJ/mol。这表明C4位置在热力学上更有利于氢原子的添加。另外,氢原子从C2位置转移到C1位置也表现出有利的活化势垒,为51.6 kJ/mol。进一步观察发现,从C4转移到C4a和从C1转移到C9a的氢转移步骤分别受到较高活化势垒的限制,分别为141.9和131.2 kJ/mol。因此,通过氢转移将2,3-二氢蒽异构化为9,10-二氢蒽具有挑战性,并且在动力学上受到“C-a位置”的阻碍。

图3 2,3-二氢蒽的氢转移异构化反应合成9,10-二氢蒽
随后,本工作讨论了2,3-二氢蒽通过分子间氢转移异构化为9,10-二氢蒽。根据图4所示,2,3-二氢蒽具有与相邻吸附的蒽分子结合的能力。具体而言,位于2,3-二氢蒽末端环内的C2位置的氢原子可能向蒽的中心环内的C9位置过渡。通过DFT计算得知,这种分子间氢转移的活化吉布斯自由能为84.3 kJ/mol,这一步骤是放热的,吉布斯自由反应能为−12.3 kJ/mol。同样地,氢原子从3-单氢蒽的C3位转移到9-单氢蒽的C10位在热力学和动力学上也是可行的。该过程的计算活化吉布斯自由能为47.6 kJ/mol,吉布斯自由反应能为−84.9 kJ/mol,表明该步骤具有高度放热性质。因此,2,3-二氢蒽通过分子间氢转移转化为9,10-二氢蒽。

图4 通过分子间氢化物转移将2,3-二氢蒽异构化为9,10-二氢蒽
我们进一步探讨了在Pt1位点和Pt1/HY位点蒽中间环开环生成BTX的反应机制。涉及C8a−C9键断裂的第一个开环步骤是Pt1位点完全蒽加氢裂化过程中的速率限制步骤,其最高吉布斯自由能垒为182.5 kJ/mol。而在双功能Pt1/BAS位点上进行的蒽完全加氢裂化中,从中心环C4a位置的BAS位点开始的质子化步骤具有最高的动力学相关性,其最高活化吉布斯自由能为107.4 kJ/mol。因此,在双功能Pt1/BAS位点上进行蒽的加氢裂化在动力学上更为可行。此外,通过晶体轨道重叠布居(COHP)分析进一步证明,相比于解离在Pt1上的H,当C4a位置接受来自BAS的H时,对C4a-C10的削弱程度更明显,因此C4a-C10更容易断键。这些结果表明,在双功能Pt1/BAS位点上的蒽加氢裂化活性更高。

图5 在Pt1位点完成蒽加氢裂化的催化循环

图6 通过中心开环路线在双功能Pt1/BAS位点完成蒽加氢裂化的催化循环
图7 C4a−C10键强度的特征在于不同蒽氢化程度的ICOHP
除了通过中心开环进行蒽加氢裂化外,我们还研究了通过双功能Pt1/BAS位点的末端开环进行的蒽加氢裂解。根据DFT计算结果如图8所示,表明通过末端开环路线进行完全蒽加氢裂化的动力学上最相关的步骤是BAS位点的第一个质子化步骤,其最高活化势垒为143.8 kJ/mol。综上所述,从动力学角度来看,优选通过双功能Pt1/BAS位点的中心开环路线进行蒽加氢裂化。DFT计算结果表明,在Pt1/HY沸石催化剂上,蒽加氢裂化的中心开环反应路线在动力学上是有利的。

图8 通过末端环开环路线在双功能Pt1/HY位点完成蒽加氢裂化的催化循环
为了比较带支链稠环芳烃开环和支链C-C键断裂的可能性,我们对一个带有C8(正辛基)蒽分子在Pt1/HY催化剂上加氢裂化反应机理进行了计算。图9和图10分别给出了辛基蒽分子在B酸位质子的作用下支链和蒽环端环连接处(端环C2)以及支链C4位置C-C断裂的反应路径。我们发现,辛基蒽分子以端环C2位置和B酸位的作用(-16.0 kJ/mol)比其辛基支链C4-C5中间位置(+5.8 kJ/mol)更强。吸附后,B酸位质子转移到辛基蒽分子生成一个正碳离子中间体(C22H27+),然后该正碳离子分解,即C-C断裂。根据质子化位置的不同,端环处支链断裂可以生成(蒽和辛烷),支链上C4-C5处断裂则生成丁烯和丁基蒽或丁烷和丁烯基蒽。其中在端环和支链结合处断裂生成辛烷和蒽在热力学和动力学上都较为有利。
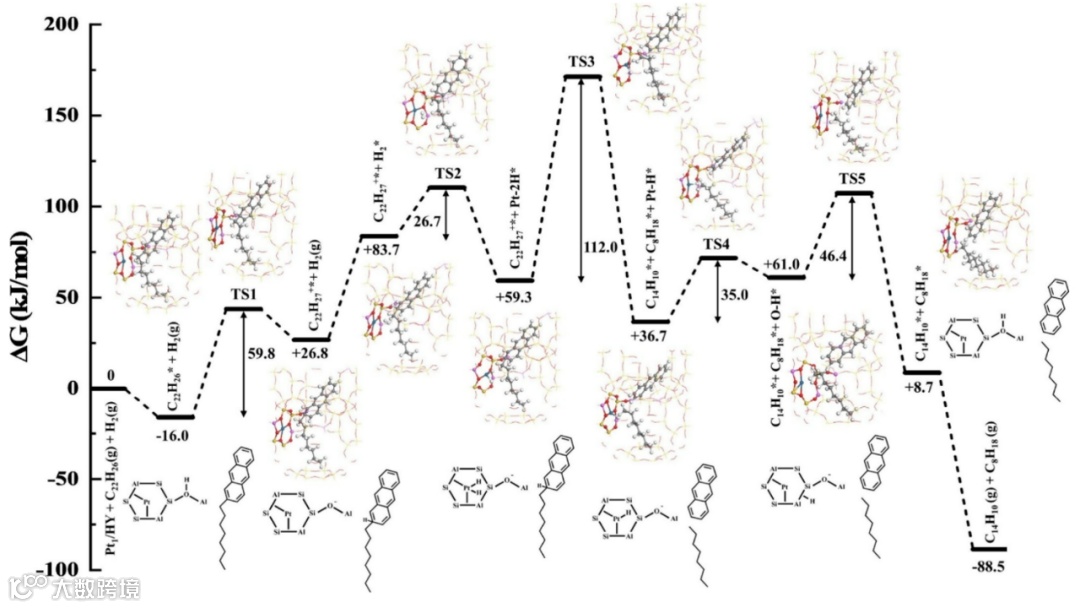
图9 DFT计算的Pt1/HY催化剂上2-辛基蒽加氢裂化为蒽和正辛烷的吉布斯自由能分布
图10 DFT计算的在Pt1/HY催化剂上2-辛基蒽加氢裂化成一对产物的吉布斯自由能分布
根据计算结果,辛基蒽在Pt1/HY催化剂上加氢裂化反应的机理被进一步探讨。图11显示,在有氢气存在的条件下,辛基蒽分子中间环的加氢开环反应能量计算结果。研究表明,辛基蒽分子的中间环开环反应至少需要2个氢气分子的参与。与蒽分子中间环开环步骤类似,该反应首先在中间环9,10位上加上两个氢原子(来自第一个氢分子在Pt1上的分解),然后B酸位的质子加到9a位置。随后,第二个氢分子在Pt上分解生成两个氢原子,其中一个氢原子加到4a位,导致C-C键断裂(即开环),其过渡态为图11中TS7,即能量最高处。研究结果表明,在带支链的稠环芳烃加氢裂解中,支链断裂相对于中间环开环反应在动力学上更为有利。换言之,在这一类反应中,端环和支链连接处的断裂相对于中间环的开环反应更容易发生。
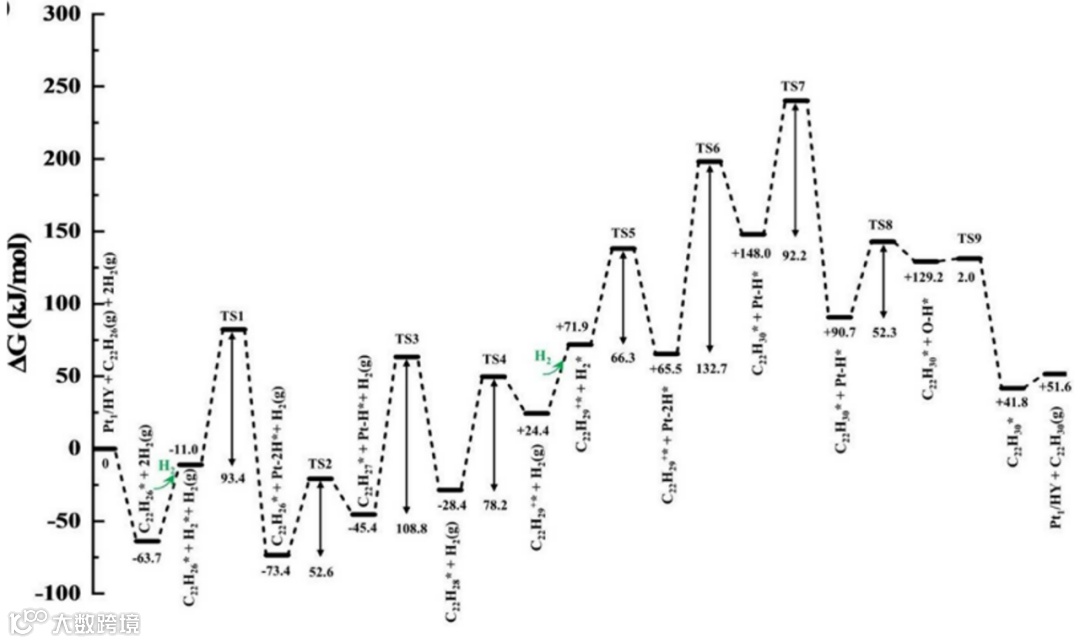
图11 DFT计算的Pt1/HY催化剂上2-辛基蒽通过中心开环加氢裂化的吉布斯自由能分布
本工作采用密度泛函理论计算,系统地研究了多环芳香烃在Pt1/HY催化剂上的加氢裂化反应机理。结果表明“C-a位”是阻止氢从末端环转移到中心环的屏障,2,3-二氢蒽异构化为9,10-二氢蒽是在附近蒽分子的辅助下可以通过分子间氢转移实现。与从Pt1位点加氢相比,在部分加氢饱和的蒽芳环上添加来自Brønsted酸性位点(BAS)的质子会明显削弱C−C键,导致中心环开环过程在动力学上是有利的。DFT结果表明,在Pt1/HY催化剂上,蒽加氢裂化的中心开环在动力学上是有利的,产生的主要产物是BTX,而不是丁苯和正丁烷。此外,还研究了以辛基蒽为探针分子的支链多环芳烃的加氢裂化反应。已经发现辛基蒽在环-支链连接位置的脱烷基化在动力学上比中心环开环和辛基链的裂解更可行。本工作对双功能HY分子筛负载金属催化剂上多环芳烃的加氢裂化反应途径提供了全面而深入的机理理解。
中国石化大连石油石化研究院柳伟和天津工业大学梅东海教授为论文的共同通讯作者,2021级博士研究生赵文茹为论文第一作者。本工作得到了中国石化大连石油石化研究院的资助。
Wenru Zhao, Hui Yu, Shaozhong Peng, Wei Liu,* Weiwei Zhang, and Donghai Mei*, Mechanistic Understanding of Anthracene Hydrocracking over HY Zeolite Encapsulated Single-Atom Pt Catalysts, ACS Catal. 2024, 14, 8836−8855.
欢迎关注我们,订阅更多最新消息 “邃瞳科学云”推出专业的自然科学直播服务啦!不仅直播团队专业,直播画面出色,而且传播渠道多,宣传效果佳。
“邃瞳科学云"平台正在收集、整理各类学术会议信息,欢迎学会、期刊、会议组织方择优在邃瞳平台上进行线上直播,希望藉此帮助广大科研人员跨越时空的限制,实现自由、畅通地交流互动。欢迎老师同学们提供会议信息(会有礼品赠送),学会、期刊、会议组织方商谈合作,均请联系潘经理:18612651915(微信同)。
投稿、荐稿、爆料:Editor@scisight.cn
“邃瞳科学云”推出专业的自然科学直播服务啦!不仅直播团队专业,直播画面出色,而且传播渠道多,宣传效果佳。
“邃瞳科学云"平台正在收集、整理各类学术会议信息,欢迎学会、期刊、会议组织方择优在邃瞳平台上进行线上直播,希望藉此帮助广大科研人员跨越时空的限制,实现自由、畅通地交流互动。欢迎老师同学们提供会议信息(会有礼品赠送),学会、期刊、会议组织方商谈合作,均请联系潘经理:18612651915(微信同)。
投稿、荐稿、爆料:Editor@scisight.cn














