

Sliding Window Interaction Grammar (SWING): a generalized interaction language model for peptide and protein interactions
滑动窗口交互语法(SWING):用于肽和蛋白质相互作用的通用交互语言模型

📑 基本信息
- 期刊
: Nature Methods - 时间
: 2025-07-28 - DOI
: 10.1038/s41592-025-02723-1 - 作者
: Jane C. Siwek*, Alisa A. Omelchenko*, Prabal Chhibbar*, Sanya Arshad, AnnaElaine Rosengart, Iliyan Nazarali, Akash Patel, Kiran Nazarali, Javad Rahimikollu, Jeremy S. Tilstra, Mark J. Shlomchik, David R. Koes, Alok V. Joglekar*, Jishnu Das* - 机构
: University of Pittsburgh School of Medicine, Center for Systems Immunology, Department of Immunology, Department of Computational and Systems Biology, Carnegie Mellon University
🔍 导读
该研究针对现有蛋白质语言模型在学习蛋白质相互作用语言方面的不足,开发了一种交互语言模型SWING,通过利用氨基酸性质差异生成交互词汇表,成功预测了I类和II类肽-主要组织相容性复合物相互作用,并能跨类别预测和准确预测变异对特定蛋白质相互作用的破坏,展现了零样本学习能力和广泛的生物学应用价值。
🧩 引言
背景与意义
蛋白质语言模型在蛋白质工程和功能预测方面取得了革命性进展,但在捕获蛋白质间相互作用方面存在根本性局限。蛋白质通过与其他蛋白质的相互作用执行功能,而现有模型主要通过分别嵌入相互作用分子然后组合嵌入来预测蛋白质-蛋白质相互作用,这种方法无法有效捕获相互作用的固有上下文特异性和对相互作用至关重要的残基接触点。
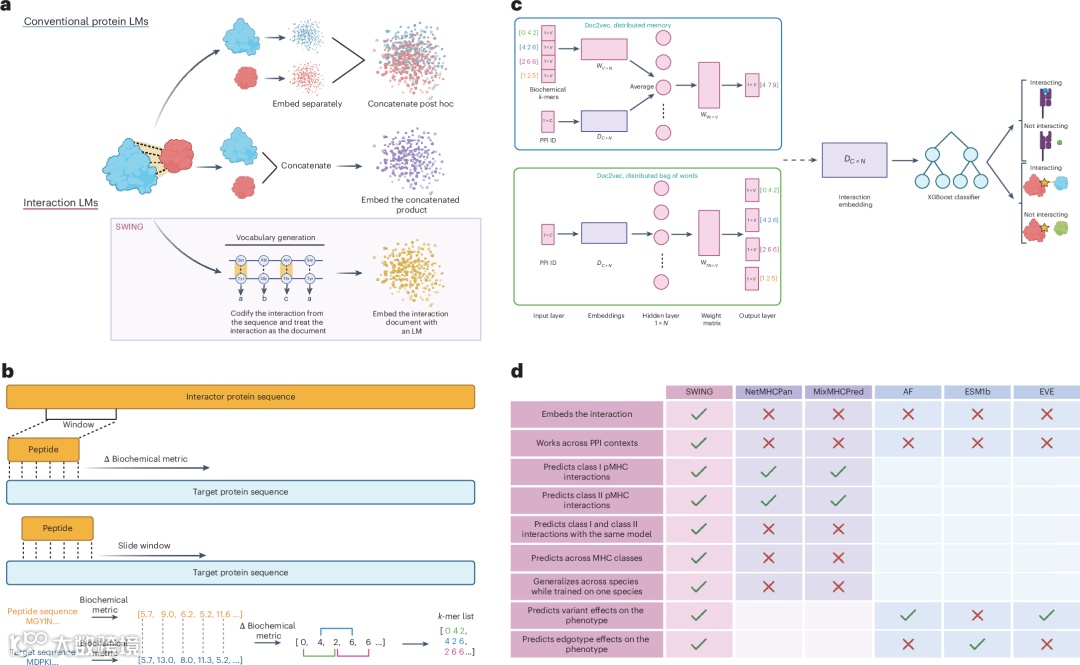
图表解读:该图展示了SWING与传统蛋白质语言模型的根本差异,突出了交互语言模型预先编码成对残基信息的创新架构,体现了从单独处理蛋白质序列到直接建模相互作用语言的范式转变。
现有不足
传统蛋白质语言模型通过聚合每个位置所有氨基酸的贡献来生成蛋白质嵌入,未考虑固有的上下文特异性或对相互作用至关重要的残基接触点。序列长度也是将蛋白质语言模型扩展到所有蛋白质类别的主要障碍,这些模型在学习蛋白质相互作用语言方面表现不佳。
问题与动机
蛋白质相互作用中高度保守的子序列促进相互作用分子之间的结合,局部信息分布在少数子序列内决定蛋白质功能。研究假设蛋白质中的局部区域在上下文优先化下应该能够捕获对相互作用重要的有意义表示,这为开发专门的交互语言模型提供了科学动机。
目标与创新
开发SWING这一长度无关的交互语言模型,能够捕获蛋白质-蛋白质或蛋白质-肽相互作用的语言。创新点包括:预嵌入编码成对残基信息、生成类似语言的表示、无需依赖现有蛋白质语言模型表示、具备零样本和迁移学习能力,能够跨不同生物学上下文进行预测。
📊 方法
数据来源与类型
使用公开可用的HLA特异性NetMHCpan v.4.1评估集和NetMHCIIpan v.4.2训练集,分别用于I类和II类pMHC相互作用数据。MHC序列从MHC限制本体论获取,选择具有质谱洗脱数据集的HLA分子,并使用MHCCluster v.2.0进行功能聚类。对于变异效应预测,使用了包含6,306个错义突变的数据集。
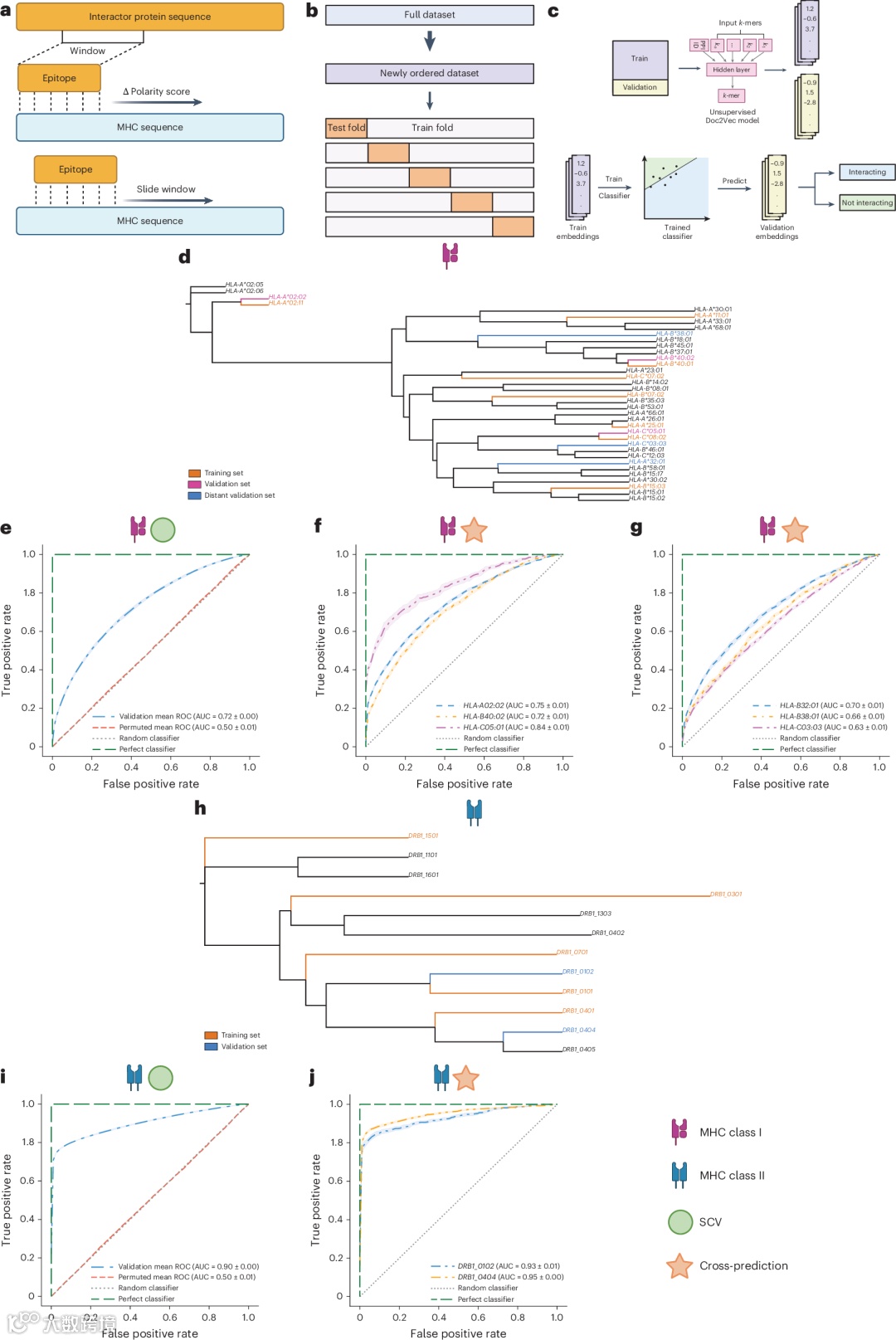
图表解读:该图展示了SWING应用于pMHC预测任务的整体框架,包括标准交叉验证和跨预测评估指标,以及I类等位基因的功能聚类分布,体现了方法的系统性评估策略。
核心方法与技术
SWING使用滑动窗口技术,将n长度肽与目标伙伴序列的n个位置完全匹配,在每个位置计算氨基酸对之间的生化差异。使用Doc2Vec模型推断相互作用嵌入,将结果分为重叠的k大小子序列作为"单词",每个相互作用作为由这些单词组成的"文档"。创新之处在于预先编码成对残基信息,无需聚合序列或嵌入。
图表解读:该图详细展示了SWING的词汇生成步骤和嵌入分类功能,包括分布式内存和分布式词袋Doc2Vec架构,突出了从生化差异编码到最终分类预测的完整技术流程。
研究流程
总体流程包括:1)语言生成-使用滑动窗口和生化指标生成交互语言;2)嵌入学习-使用Doc2Vec模型生成低维表示;3)监督学习-使用XGBoost等分类器进行下游预测任务。关键步骤包括超参数优化、交叉验证评估、跨预测性能测试,确保模型的泛化能力和生物学相关性。
📈 结果
pMHC相互作用预测性能
SWING在I类pMHC相互作用预测中表现出色,交叉验证AUC达到0.72(P < 0.001),对未见等位基因的预测准确性保持在0.72-0.84之间。对于功能上不同的未见HLA-I等位基因,AUC为0.63-0.70。II类模型在交叉验证中AUC达到0.90(P < 0.001),对未见功能不同的II类等位基因预测AUC为0.93-0.95,证明了SWING学习I类和II类pMHC相互作用语言的能力。
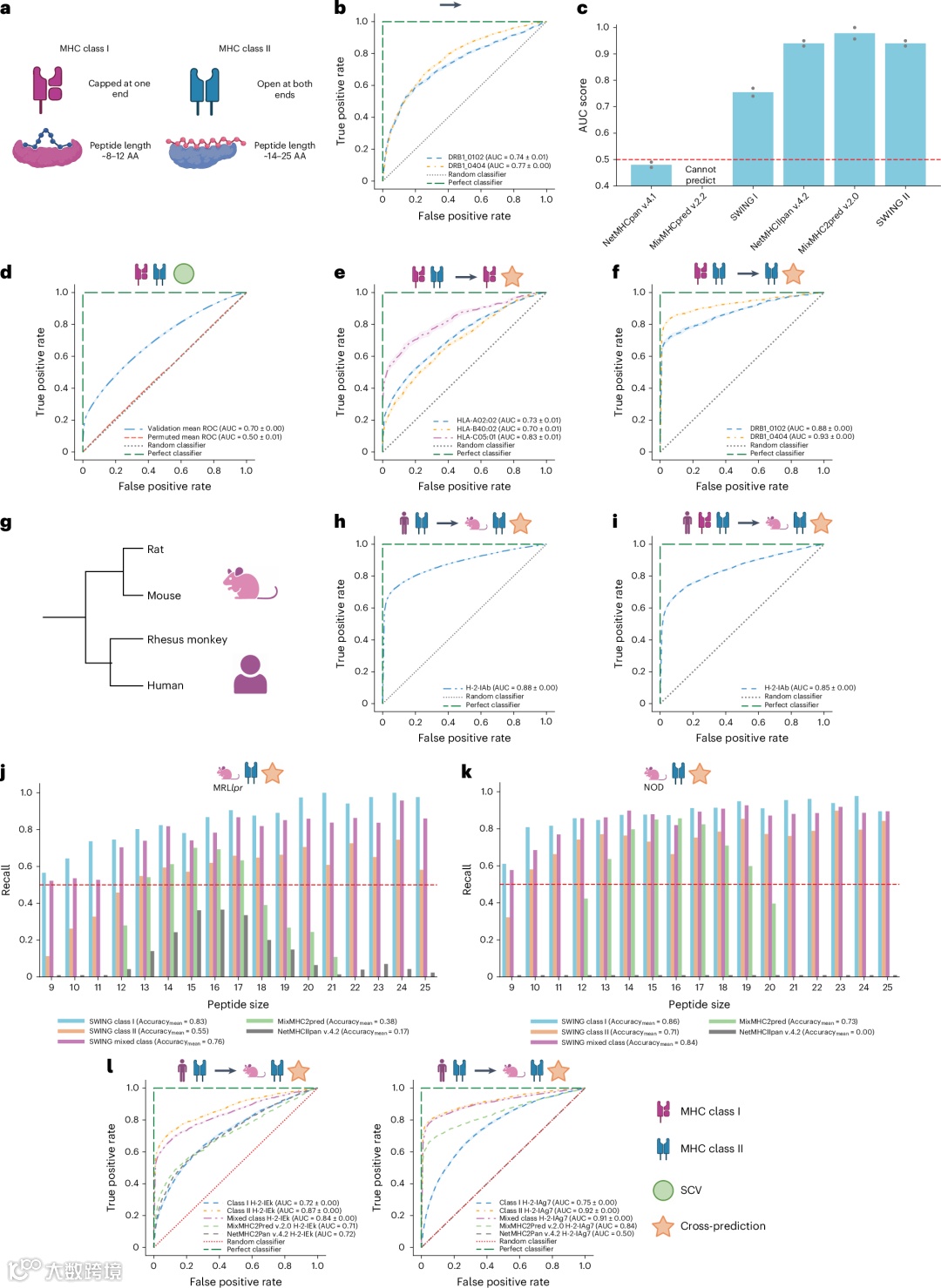
图表解读:该图展示了SWING在不同MHC类别间的零样本预测能力,包括I类模型预测II类相互作用的性能,以及与现有方法的比较结果,体现了SWING独特的跨类别预测优势。
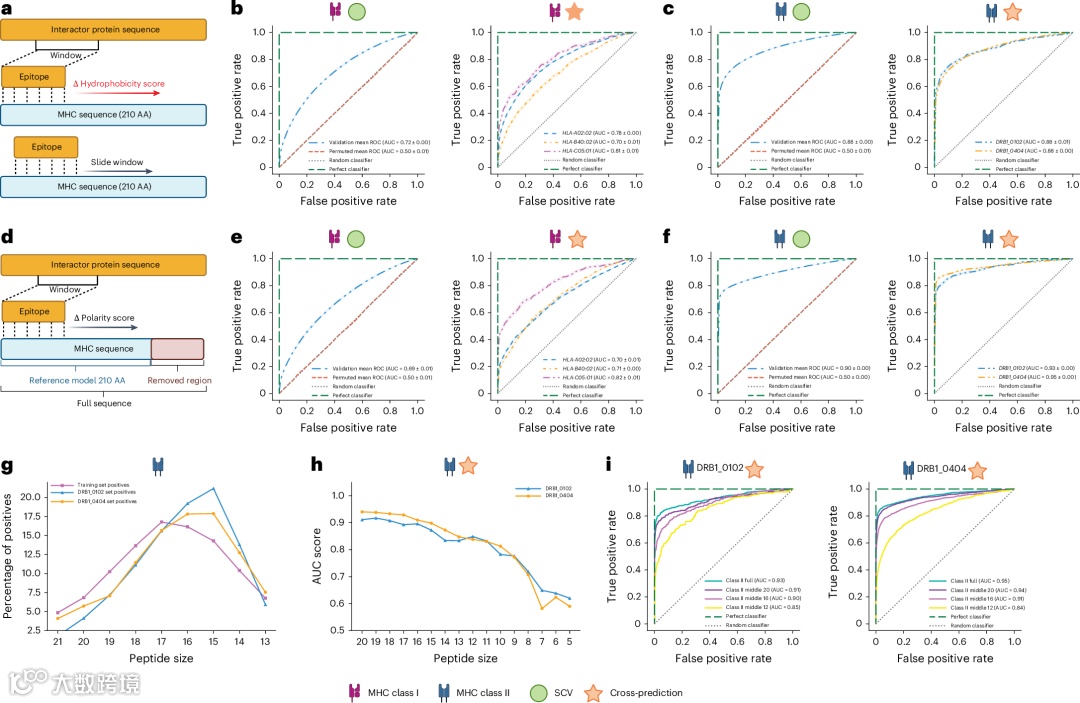
跨物种和跨类别预测能力
SWING展现了卓越的迁移学习能力,I类模型能够自信地预测II类pMHC相互作用(AUC = 0.74-0.77),这是现有方法未尝试的复杂预测任务。联合模型(同时训练I类和II类数据)能够预测两类MHC相互作用。人类SWING模型能够零样本预测小鼠pMHC相互作用,II类模型AUC达到0.88,联合模型AUC为0.85,证明了跨物种的泛化能力。
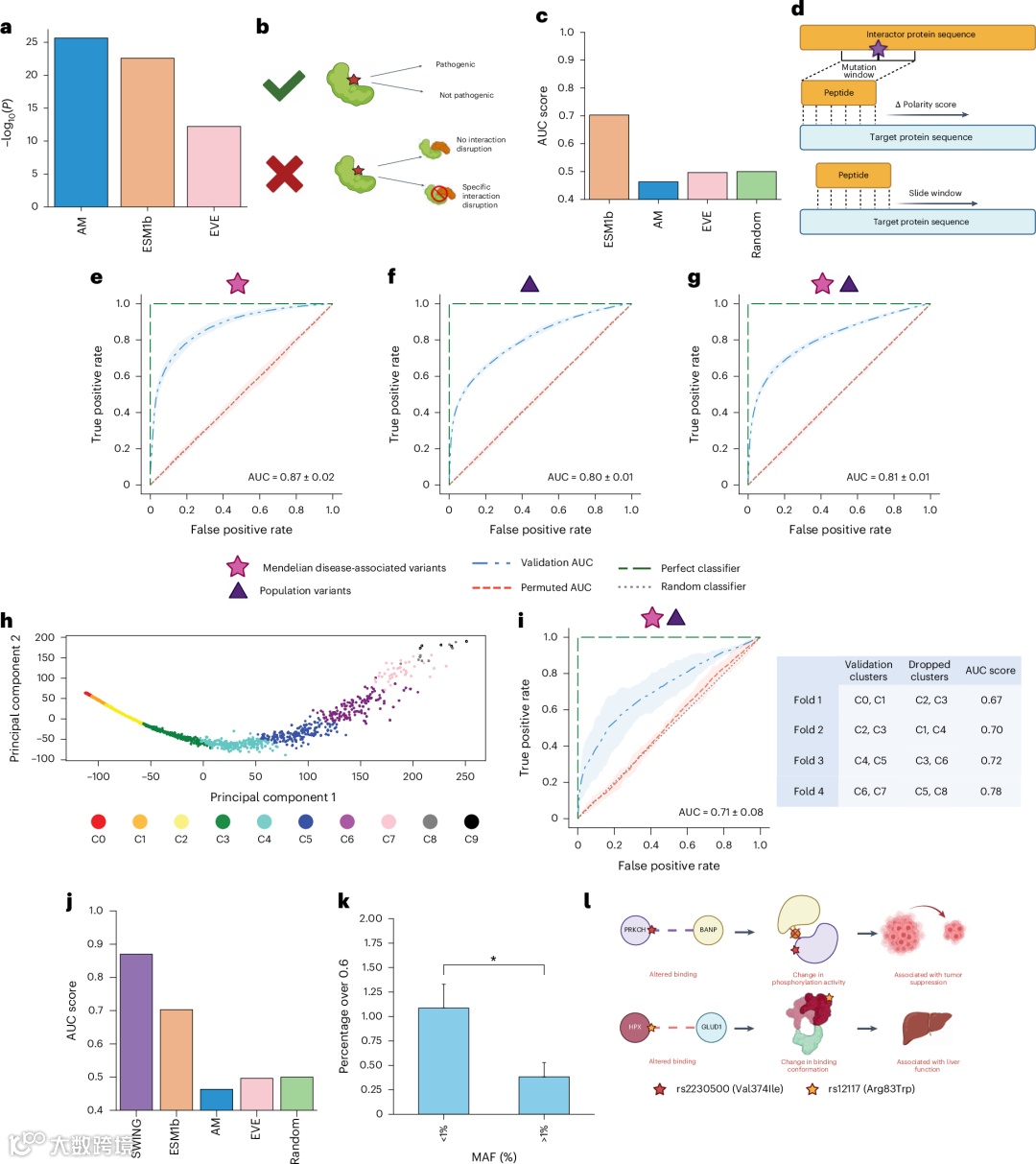
图表解读:该图展示了SWING在预测相互作用破坏方面的性能,包括与变异效应预测工具的比较,以及在不同变异类型和等位基因频率下的预测准确性,突出了SWING在变异效应预测方面的独特优势。
变异对蛋白质相互作用的破坏预测
SWING成功预测了错义突变对蛋白质相互作用的破坏效应。对于孟德尔疾病变异,预测准确性AUC达到0.87(P < 0.0001);对于群体变异,AUC为0.80(P < 0.0001);混合模型AUC为0.81(P < 0.0001)。SWING超越了更复杂的变异效应预测模型,能够预测特定相互作用的表型效应,而不仅仅是整体病理性。模型还准确预测了稀有变异比常见变异更可能破坏相互作用的生物学规律。
💡 结论
主要贡献
方法学创新:开发了首个专门用于蛋白质相互作用的语言模型SWING,通过预嵌入编码成对残基信息,克服了传统蛋白质语言模型在相互作用建模方面的局限性,为相互作用预测提供了全新的计算框架。
跨领域预测能力:SWING展现了卓越的零样本和迁移学习能力,能够在MHC类别间、物种间进行准确预测,特别是I类模型预测II类相互作用的独特能力,为数据稀缺情况下的相互作用预测提供了有效解决方案。
局限性与展望
当前SWING主要专注于蛋白质-肽相互作用预测,需要根据生物学上下文进行定制化训练,不是通用的生物相互作用基础模型。未来版本将扩展到处理两个以上输入序列,预测T细胞受体-pMHC等复杂相互作用,并处理DNA、RNA和小分子等不同分子实体,以更好地理解转录因子-DNA复合物等异质相互作用和调控网络。
想做自己的垂直领域大数据研究(电池、陶瓷、金属、医药、蛋白、环境、生态、材料、农学、地质等),可使用类似方式完成定制化数据集搜集搭建,TAG-HUB也可提供定制化科研数据集挖掘服务。
