

我们进一步将I½型和II型拮抗剂细分为A和B两个亚型。A型药物延伸过守门残基进入背部凹槽。相反,B型药物不延伸进入背部凹槽。根据不完整的数据表明,这种差异的可能意义是A型拮抗剂与其酶靶标结合的停留时间比B型阻断剂更长。

图1. Type I和TypeII结合位点

小分子蛋白激酶抑制剂(PKIs)的未来前景是充满希望的。随着新药的不断开发,大约有110种新型激酶处于临床试验阶段,这表明这一领域仍有很大的未开发潜力。未来的研究将集中在克服药物抗性、优化药代动力学和药效学特性以及个性化治疗上。联合疗法也将成为提高疗效的重要策略,通过将PKIs与其他治疗方法结合,可能有效地解决药物抗性问题。

图3. FDA批准的蛋白激酶抑制剂
ICE拥有用于筛选各种类型抑制剂的蛋白质,并且提供相应的筛选服务。进入下面的链接或通过我们的网站发送消息来了解更多信息:https://protein.ice-biosci.com/
ABL抑制剂



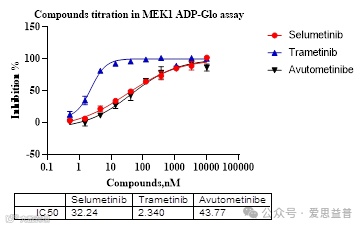
MEK1抑制剂

RAF/MEK/ERK通路是控制细胞生理的核心,其失调与许多癌症有关。因此,构成这一途径的蛋白一直受到强烈的药物发现和开发努力的影响。变构MEK抑制剂(Allosteric MEK inhibitors, MEKi)对RAF/ MEK/ERK通路信号通路具有复杂的作用,在临床上与BRAF抑制剂联合应用于恶性黑色素瘤。变构MEKi可以通过至少四种不同机制中的一种或多种来阻断RAF/MEK/ERK信号通路的激活:1)通过与uMEK结合并诱导阻止其与RAF结合;2)允许uMEK与RAF结合,使MEK稳定于抗磷酸化构象;3)通过阻止RAF释放pMEK,阻止ERK磷酸化;4)抑制游离pMEK以阻断其磷酸化ERK的能力。研究结果表明,MEK抑制剂除抑制MEK外还对BRAF/MEK复合物起抑制作用。

图6. MEK1变构抑制剂结合位点
EGFR抑制剂

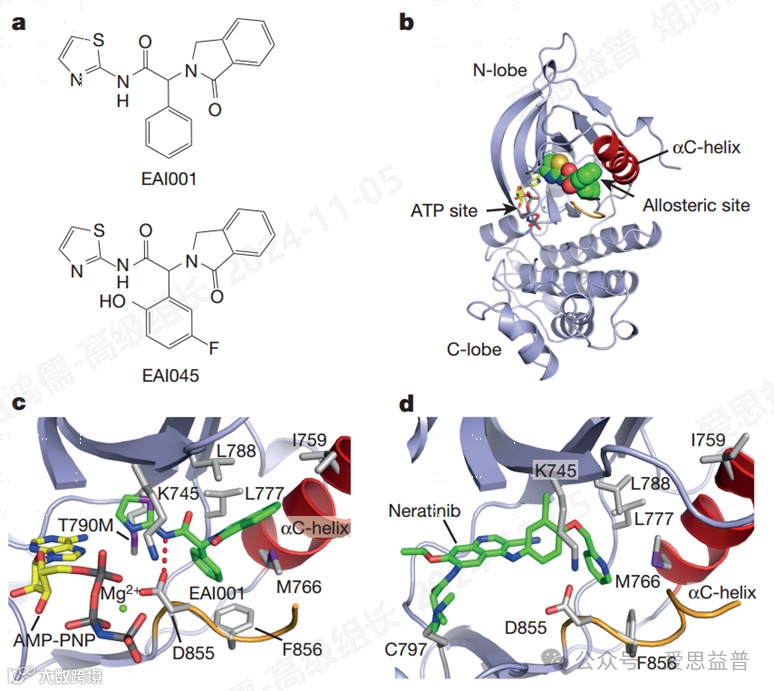

图9. Afatinib和Osimertinib 为EGFR共价抑制剂。BLU-945 为I型抑制剂,EAI-045是III抑制剂。
AKT抑制剂

最新的研究表明,在没有上游信号的情况下,AKT的PH结构域和激酶结构域之间的相互作用使其保持在一种自抑制的闭合状态,这阻止了PDK1对T308位点的磷酸化。而当接收到上游信号时,AKT会从这种自抑制状态转变为开放状态,使得磷酸化和激活成为可能。研究者通过对大量人类肿瘤样本的分析发现,PH-KD接触位点的突变在癌症中是存在的,这种突变破坏了PH和KD之间的相互作用,可能是AKT在癌症中激活的一种新机制。更重要的是,这些肿瘤特异性的突变不仅能够激活AKT,还可能改变肿瘤对变构AKT抑制剂的敏感性,这对于开发新的癌症治疗策略具有重要意义。

图10. ATK两种不同激活方式
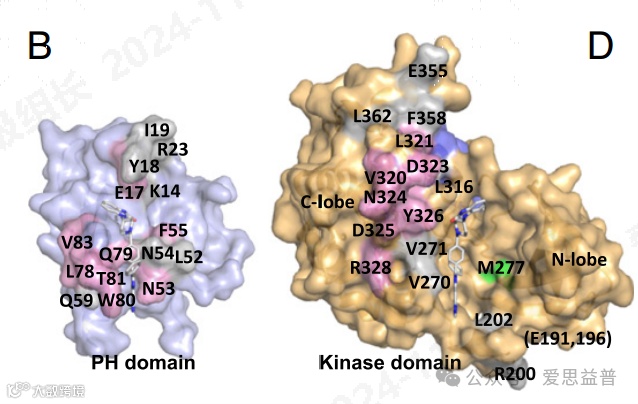
图11. ATK变构抑制剂结合位点

JAK抑制剂


图13. BMS-986165与TYK2 JH2共晶结构

图14. BMS-986165为TYK2 VI 型抑制剂
BTK抑制剂

尽管大多数CLL (chronic lymphocytic leukemia) 患者对单药治疗 BTK 抑制剂有多年的反应,但需要持续治疗,并且最终会发展出 BTK 或 PLCγ2 的耐药突变。共价抑制剂特别容易受到 C481 突变的影响,因为它们依赖于与该残基的结合。非共价 BTK 抑制剂克服了这一限制,但仍易受到新型 BTK 激酶结构域突变的影响。BTK L528W、V416L 和 A428D 被归类为激酶功能受损的耐药突变,而 BTK C481S 和 T474I 被归类为激酶功能完整突变。BTK 降解剂仍在进行临床试验,但早期结果表明它们可以克服激酶受损和激酶熟练的获得性 BTK 耐药突变,但尚不清楚它们是否可以靶向下游 PLCγ2 激活突变。
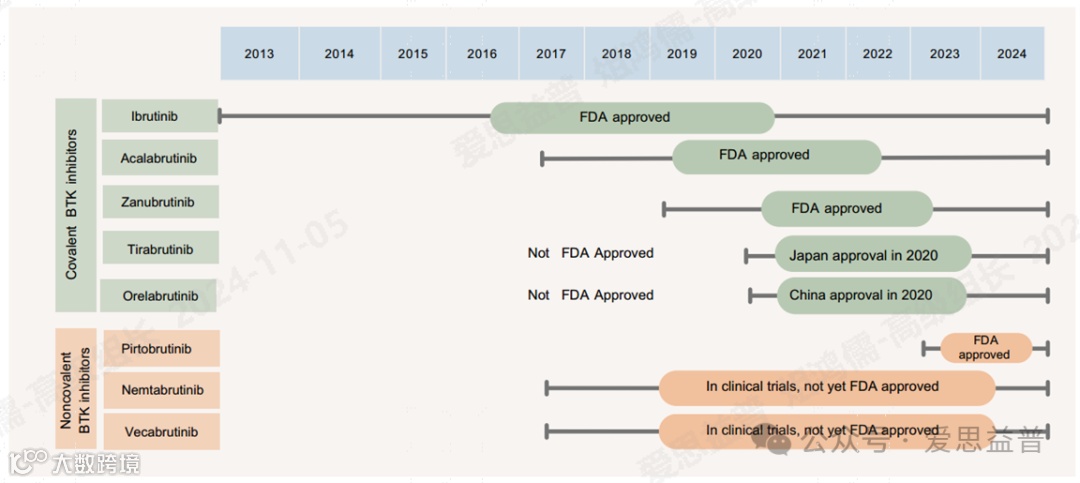
图15. FDA批准的BTK抑制剂

图16. Ibrutinib和Acalabrutinib 为BTK共价抑制剂,Pirtobrutinib为BTK I型抑制剂

公司介绍
