

PLK1(Polo-like Kinase 1,PLK1)以黑腹果蝇的polo基因命名,是一种丝氨酸/苏氨酸蛋白激酶,广泛的存在于真核细胞中,参与有丝分裂的起始、维持和结束等进程。人源PLK1家族成员有:PLK1、PLK12、PLK13、PLK14和PLK15。其中PLK1被广泛研究。诸多研究表明,PLK1在多种癌症中高表达,是肿瘤的快速增殖所必须的,且在一些癌症中,常与较差的预后有关,推断认为PLK1作为致癌基因加快肿瘤的发生和发展。此外,最近的研究表明,PLK1 可能具有其他重要功能,例如调节DNA 合成、细胞凋亡、p53 反式激活、DNA 损伤检查点恢复和驱动肿瘤细胞转移等[1]。因此,PLK1也被认为是治疗癌症的潜在的治疗靶点,一些靶向PLK1的抑制剂和siRNA已经用于临床试验中。
1.PLK1的结构
PLK1蛋白是由一个高度保守的N端激酶催化结构域(kinase domain,KD)、C端2个polo盒样结构域(polo-box domain,PBD),PB1和PB2,以及中间的连接区域组成(图1)。PLK1的激酶结构域有核定位信号区(NLS)、有丝分裂结束后的破坏盒(D-box)、一段端粒环(T-loop),与ATP的结合和酶活性密切相关[2]。正常情况下,PLK1的KD和PBD结合,会抑制激酶中Thr210的磷酸化,进而抑制PLK1的激酶活性,属于一种自抑制机制[3]。当PBD结构域和某些蛋白的磷酸肽结合,被招募到特定的细胞位置,KD被释放,与激酶结构域的T-loop分离,PLK1的激酶活性被激活[4]。

图1.1 PLK1的结构图
(图1.1来自Liu Z, Sun Q, Wang X. PLK1, A Potential Target for Cancer Therapy. Transl Oncol. 2017 Feb;10(1):22-32.)
上皮-间质转化(epithelial-to-mesenchymal transition,EMT)在很多癌症的发展中发挥着至关重要的作用,包括癌细胞的迁移、侵袭、血管生成和耐药等。EMT主要通过上皮细胞标志物cytokeratins和E-cadherin的缺失和间充质细胞标志物N-cadherin、vimentin和纤连蛋白的上调等指标进行检测,这一转化过程会导致细胞之间的粘附性降低[5]。杂合EMT的主要特征是肿瘤细胞中上皮基因和间质基因的共同表达,促进癌症的发生、转移和浸润[6]。

EMT的发生发展与MEK1/2-ERK1/2-Fra1-ZEB1/2信号通路的激活成正相关,Wu[7]等人研究了MEK1/2-ERK1/2-Fra1-ZEB1/2的上游调控因子CRAF与PLK1的互作在EMT过程发挥的重要作用。PLK1直接磷酸化S338和S339位点的CRAF,激活CRAF;活化的CRAF经过S621位点的自磷酸化,阻止了蛋白酶介导的CRAF降解,CRAF的稳定也在一定程度上增加了PLK1的磷酸化,进而产生了一个正反馈的循环。该研究将过表达PLK1的前列腺上皮细胞RWPE-1移植到小鼠体内,在NSG小鼠中RWPE-1-PLK1表现出致瘤性和促使肿瘤转移的能力。

图2.1.2 PLK1在RWPE-1细胞中的异位表达的致瘤性和肺部微转移[7]
肺癌已经成为全球主要致死癌症之一,约40%的肺癌患者都会发生肿瘤转移,转移至脑、肝和肾上腺等部位,严重威胁人的生命[8]。非小细胞肺癌 (NSCLC) 患者约占所有肺癌的 85%,生存率极低,临床相关性结果表明,PLK1和TNFAIP6是转移性 NSCLC 患者生存率低的强预测因子。研究[9]发现在T210位点磷酸化的PLK1能促进癌细胞的迁移、侵袭和致瘤性,而PLK1活性的丧失阻止了肿瘤发生和转移活性,S137磷酸化的PLK1则不具有该特性。T210- PLK1 的表达激活 了TGF-β 信号通路并上调TNFAIP6,从而促进 NSCLC 的转移和侵袭。因此,PLK1 和 TSG6 是转移性非小细胞肺癌治疗的有价值的治疗靶点。PLK1促进EMT的作用同样在胃癌细胞系SGC7901和MKN28中得到验证,并得出PLK1可能通过AKT信号通路促进胃癌的转移[10,11]。
2.2 细胞凋亡
上皮-间质转化发生在相当一部分 NSCLC 病例(≥20%)中,可介导癌基因成瘾、表皮生长因子受体抑制剂耐药和化疗耐药的丧失。PLK1 抑制导致 G2/M 期阻滞,但只有治疗敏感的细胞系在 PLK1 抑制后发生大量凋亡。具有高上皮-间质转化基因特征评分的 NSCLC 细胞系(间充质细胞系)对 PLK1 抑制的敏感性高于上皮细胞系;蛋白质组学分析结果得到相同的结论[12]。通过表达 miR-200 诱导上皮表型增加了细胞对 PLK1 抑制的抗性。该研究将肿瘤分为上皮细胞系和间充质细胞系两种亚型,针对不同亚型对PLK1抑制剂的敏感性,对肿瘤进行治疗,在临床应用上具有重要意义。Caspase-9是参与细胞凋亡的关键酶。Plk1的表达增加了pro-Caspase9和pro-Caspase3的水平,抑制了细胞凋亡,表明Plk1具有抑制细胞凋亡的作用[13]。
焦亡被称为炎症引起的细胞程序性死亡,属于凋亡的一种,在近年来受到诸多关注,NLRP3、GSDMD、AIM2、NLRC4、NLRP1等是焦亡研究中最受关注的位点。最近,研究发现PLK1抑制剂被证实是一种有效的治疗食管鳞状细胞癌(ESCC)的方法,BI2536 通过激活caspase-3,诱导GSDME高表达细胞系中GSDME裂解为C-末端片段 (GSDME-CT) 和 N-端片段 (GSDME-NT),GSDME-NT 也参与膜孔的形成,导致细胞焦亡,进而增强ESCC细胞系对顺铂(DDP)的敏感性[14]。BI2536还可以通过促进caspase-8介导的凋亡通路的激活和PARP的激活来增加ESCC细胞系的凋亡,然而,BI2536对其他已知引起细胞凋亡的生化标记,如caspase-9、cytochromeC和自噬,并无显著影响。

2.2.1 BI2536和DDP联合用药活化Caspase-3和增加DSDME胞质中的累积进而诱导焦亡[14]
PLK1调控细胞周期,调控细胞增殖,而敲低PLK1可显著抑制胶质瘤细胞U87和U251的增殖、迁移、侵袭,诱导细胞凋亡。此外,数据显示,PLK1的下调显著提高了U251和U87细胞中cleaved caspase-3、BIM、BAX和E-cadherin的表达,降低了MMP9、ATG5和LC3-II的表达[15]。靶向PLK1可能与自噬的调控有关。在对吉非替尼的抗癌研究中发现,肝脏特异性Atg7+/-杂合子小鼠的肝损伤比正常对照小鼠更轻,表明自噬参与了吉非替尼促进的肝毒性。而这种自噬促进的细胞凋亡依赖于PLK1 ,AAV-8介导的PLK1下调和PLK1抑制剂BI-2536都可以通过消除COX6A1蛋白的自噬降解来减轻吉非替尼诱导的肝毒性。此外,PLK1抑制不影响吉非替尼的抗癌活性[16]。PLK1与mTOR的相互作用也与自噬密切相关[17,18]。含有mSin1.5的mTORC2被认为在应激信号传递中具有重要功能,PLK1的Rictor磷酸化导致mSin1.5水平的增加,因此在调控该通路中也可能需要Rictor-S1162的PLK1磷酸化[19] 。研究发现,抑制PLK1可导致食管鳞癌和AML中mTOR活性的衰减[20,21]。相比之下,Ruf和他的同事描述了PLK1抑制HeLa细胞中的mTORC1,从而调节营养饥饿条件下的自噬[22]。
2.3 DNA损伤
人类基因组在各种外源性因素和内源性因素破坏造成不同类型的DNA损伤,包括DNA单链断裂(SSB)、DNA双链断裂(DSB)和复制叉崩溃等。Rad51重组酶以细胞周期和DNA损伤反应的方式被Plk1在S14处直接磷酸化,随后刺激CK2介导的T13磷酸化, CK2对Rad51的T13磷酸化触发与MRN组分Nbs1的FHA结构域的直接作用,S14或T13处的Rad51磷酸化对于准确的HR以及细胞对IR和PPAR抑制的抵抗力中发挥着重要作用[23]。研究发现DSB修复因子CtIP是由CDK1/Aurora A和PLK1共同磷酸化的[24]。CDK1/Aurora A介导的CtIP在丝氨酸327处的磷酸化触发了CtIP与PLK1 polo-box结构域的结合,进而促进PLK1在丝氨酸723处磷酸化CtIP。PLK1磷酸化的CtIP突变体不能启动扩展末端切除,因此不能介导同源重组和G2/M检查点,但可以介导MMEJ。这些数据暗示PLK1可能以CtIP为靶点,促进易出错的MMEJ,并使G2/M检查点失活[25]。PLK3介导的CtIP磷酸化促进了CtIP-BRCA1相互作用,从而启动G1期的末端切除和非同源末端连接(NHEJ)[26]。

2.3 PLK1在DNA损伤修复中的作用[23]
3.PLK1抑制剂在癌症治疗中的研究和应用
3.1 BI2536
Martin Steegmaier[27]报道了一种有效的哺乳动物PLK1小分子抑制剂BI 2536,BI 2536是第一种能诱导PLK1抑制的所有特征的强效和选择性PLK1抑制剂,它能在低纳摩尔浓度下抑制PLK1酶的活性。该化合、物可在具有不同组织来源和肿瘤基因组特征的人类癌细胞系中有效地引起有丝分裂停止和诱导凋亡,细胞在前中期停止,积累磷酸组蛋白H3,并包含异常的有丝分裂纺锤体。同时,BI 2536在耐受良好的静脉剂量方案下,抑制裸小鼠人肿瘤异种移植瘤生长,诱导大肿瘤消退。

图3.1 BI2536的结构图
(图3.1来自https://www.chemsrc.com/cas/755038-02-9_122800.html)
三阴性乳腺癌 (TNBC)的高发归因于肿瘤起始细胞 (TICs)的存在,通过全基因组人类激酶小干扰 RNA (siRNA) 文库筛选,发现PLK1可能成为治疗TNBC 的潜在靶点。siRNA 或 BI 2536 抑制 PLK1 可阻止 TNBC 的生长,包括 CD44高/CD24 -/低TIC 亚群和乳腺球形成[28]。BI2536在肾上腺皮质癌[29]、前列腺癌[30]等也显示出令人兴奋的结果。在抗纤维化方面,BI2536也表现出其优势[31]。BI 2536 不会对心肌细胞产生不利影响,但会严重影响原代成纤维细胞。成纤维细胞在具有单极纺锤体的有丝分裂中被停滞,并在长时间停滞后死亡。在 BI 2536 存在下,内皮素-1 和去氧肾上腺素刺激对心肌细胞和肥大反应没有观察到影响。
3.2 BI6727(volasertib)
BI6727,分子式C34H50N8O3,是一种二氢蝶啶酮类化合物的 ATP 竞争性激酶抑制剂,IC50为0.87 nM,在 BI 2536 二氢蝶啶酮的结构基础上改良的。Volasertib选择性抑制PLK1,在多种肿瘤细胞中诱导选择性G2/M阻滞和凋亡,而在正常细胞中引起G1和G2期可逆的细胞阻滞而不发生凋亡。
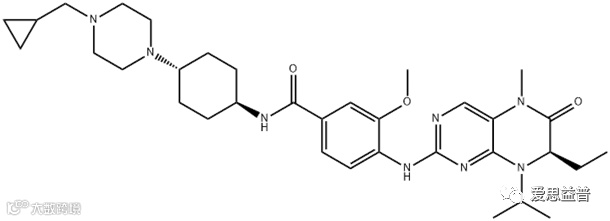
图3.2.1 BI6727的结构图
(图3.2.1来自https://www.chemicalbook.com/ChemicalProductProperty_CN_CB12501403.htm)
早在2009年,就有研究表明,通过免疫荧光显微镜和DNA 含量的荧光细胞分选分析BI6727对NCI-H460细胞增殖的抑制,在24 h,细胞停滞在G2-M期,结合细胞增殖结果,利用PARP的蛋白表达结果确定BI6727在48h诱导NCI-H460细胞的凋亡[32]。上述结果在膀胱癌研究中也得到证实[33]。体内实验结果也表明,BI 6727能够抑制对高浓度长春新碱耐药的NCI-H460细胞和急性髓性白血病 (AML) 细胞系的增殖,在异位表达MDR1基因的黑色素瘤细胞系(BROmdr)中也得到了类似的结果,显示出BI 6727对这些耐药机制的低敏感性[34]。BI 6727的药代动力学特征有利于肿瘤组织在小鼠和大鼠中持续暴露,具有较高的分布量和较长的终端半衰期。BI 6727在多种癌症模型显示出显著的抗肿瘤活性,包括紫杉烷耐药结直肠癌模型等[34]。迄今为止,BI 6727可以说是体外和体内最有效的 PLK1 抑制剂之一,已在 I 期和 II 期的试验中显示出治疗潜力,并正在进行 III 期试验。该抑制剂也已尝试与其他抑制剂联合使用,并取得了令人鼓舞的成功[35]。FDA授予PLK1抑制剂volasertib突破性治疗称号,当其与低剂量阿糖胞苷(LDAC)联合使用时,可用于不适合强化缓解诱导治疗的患者对抗急性髓系白血病(AML) [36]

图3.2.2 BI6727对NCI-H460细胞的抑制和诱导其凋亡[32]
4. PLK1抑制剂联合用药的研究
单一疗法目前存在一定的缺陷:1、由于患者之间表观遗传差异,对某些患者有效的药物可能对其他患者无效;2、在肿瘤内,只有一小部分肿瘤细胞可能对药物敏感,导致肿瘤不完全破坏;3、单一药物作用会提高药物的耐药性。而针对单一疗法现有的治疗缺陷,联合疗法提供了以下策略:1、靶向两种以上的途径,以便消除具有不同遗传背景的癌细胞;2、具有优于或者协同的抗癌作用,从而在细胞产生耐药性之前迅速将其消除[37]。
和其他抗癌药一样,BI2536出现了耐药性,Solanes-Casado S[38]等人在体外建立了耐BI2536的结直肠癌细胞系HT29R、RKOR、SW837R和HCT116R,研究耐药细胞的AXL途径、EMT和MDR1,并在RKOR中发现PLK1基因突变R136G,与耐药性密切相关。Stehle等人证明,在降低横纹肌肉瘤(RMS)细胞的细胞活力和集落形成能力方面,艾日布林(一种新型微管干扰药物)与PLK1抑制剂BI2536的共同治疗明显比单一疗法更有效[39]。另一项研究测试了BI2536和诺考达唑的联合用药,诺考达唑是一种微管毒物,可在DU145晚期前列腺癌(PCa)细胞中造成M期阻滞。这种组合治疗能协同诱导癌细胞凋亡并抑制PCa细胞的活力,但不能抑制正常的前列腺上皮细胞[40]。HBCx-10模型是人类三阴性乳腺癌(TNBC)衍生的异种移植模型,在HBCx-10模型中,阿霉素和环磷酰胺与BI2536的组合可阻止肿瘤复发[41]。Lian等人评估了BI2536与顺铂联合治疗胃癌,结果表明,BI2536增强顺铂诱导的对顺铂耐药胃癌细胞(SGC-7901/DDP)细胞活力和侵袭的抑制作用[42]。BI2536显著提高了DNA烷化剂替莫唑胺(TMZ)在体外以及使用异柠檬酸脱氢酶1(IDH1)突变星形胶质细胞的异种移植小鼠模型中的功效[43]。
结语
北京爱思益普生物科技股份有限公司(ICE)专注于从先导化合物筛选,优化到临床前候选分子阶段基于细胞和生化的药物体外筛选技术和早期药物机理研究,致力于建立全面的靶点筛选和体外生物学研究平台。
在表观遗传学靶点的平台拓展和方法研究方面,爱思益普研发团队积级开展了靶向治疗中PLK1靶点的研究和分析,已经通过酶学实验研究了BI6727对PLK1激酶活性的作用。
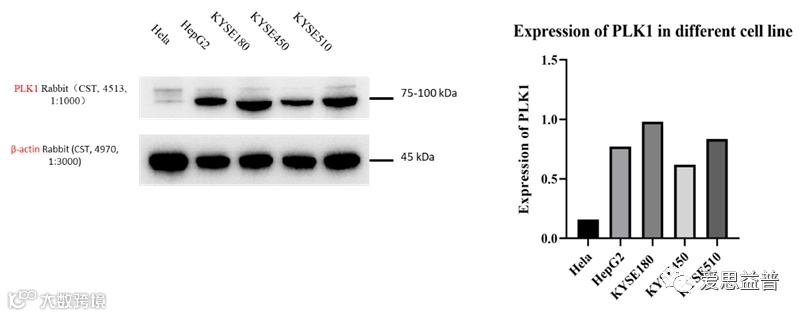

爱思益普还可通过In-cell-Western、 HCS、Western Blot、qPCR、LC-MS等技术精确精准分析PLK1蛋白靶点、突变位点和联合用药。工作内容包括前期细胞培养、细胞生长曲线测定、目的化合物筛选等。ICE公司细胞种类贮备充足、设备前沿、理论知识充分,在实验中积累了大量的研发经验,建立体外酶学、细胞、DMPK、体内一体化平台。为后期PLK1靶向药物的开发及优化奠定良好基础。
摘要
[1] Song B, Liu XS, Rice SJ, et al. Plk1 phosphorylation of orc2 and hbo1 contributes to gemcitabine resistance in pancreatic cancer. Mol Cancer Ther. 2013: 12(1): 58-68.
[2]de Carcer G, Manning G, Malumbres M. From PLK1 to PLK15: functional evolution of polo-like kinases. Cell Cycle. 2011: 10(14):2255–2262.
[3] Liu Z, Sun Q, Wang X. PLK1, A Potential Target for Cancer Therapy. Transl Oncol. 2017: 10(1): 22-32.
[4] Lowery D.M., Lim D., Yaffe M.B. Structure and function of polo-like kinases. Oncogene. 2005: 24(2): 248–259.
[5] Banyard J, Bielenberg DR. The role of EMT and MET in cancer dissemination. Connect Tissue Res. 2015: 56(5): 403-13.
[6]Pastushenko I, Mauri F, Song Y, et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature. 2021: 589(7842): 448-455.
[7] Wu Jianguo, Ivanov Andrei I, Fisher Paul B.et al. Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling [J]. Elife, 2016: 5.
[8] Tamura T, Kurishima K, Nakazawa K, Kagohashi K, Ishikawa H, Satoh H, et al. Specific organ metastases and survival in metastatic non-small-cell lung cancer. Mol Clin Oncol. 2015: 3: 217–21.
[9] Shin SB, Jang HR, Xu R, Won JY, Yim H. Active PLK1-driven metastasis is amplified by TGF-β signaling that forms a positive feedback loop in non-small cell lung cancer. Oncogene. 2020: 39(4): 767-785.
10] Cai XP, Chen LD, Song HB, Zhang CX, Yuan ZW, Xiang ZX. PLK1 promotes epithelial-mesenchymal transition and metastasis of gastric carcinoma cells. Am J Transl Res. 2016: 15: 8(10): 4172-4183.
[11] Song R, Hou G, Yang J, Yuan J, Wang C, Chai T, Liu Z. Effects of PLK1 on proliferation, invasion and metastasis of gastric cancer cells through epithelial-mesenchymal transition. Oncol Lett. 2018: 16(5): 5739-5744.
[12] Ferrarotto R, Goonatilake R, Yoo SY, et al. Epithelial-Mesenchymal Transition Predicts Polo-Like Kinase 1 Inhibitor-Mediated Apoptosis in Non-Small Cell Lung Cancer. Clin Cancer Res. 2016: 22(7): 1674-1686.
[13] Liao G, Wang R, Tang DD. Plk1 Regulates Caspase-9 Phosphorylation at Ser-196 and Apoptosis of Human Airway Smooth Muscle Cells. Am J Respir Cell Mol Biol. 2022: 66(2): 223-234.
[14] Wu M, Wang Y, Yang D, et al. A PLK1 kinase inhibitor enhances the chemosensitivity of cisplatin by inducing pyroptosis in oesophageal squamous cell carcinoma. EBioMedicine. 2019: 41: 244-255.
[15] Wu ZY, Wei N. Knockdown of PLK1 inhibits invasion and promotes apoptosis in glioma cells through regulating autophagy. Eur Rev Med Pharmacol Sci. 2018: 22(9): 2723-2733.
[16] Luo P, Yan H, Du J, et al. PLK1 (polo like kinase 1)-dependent autophagy facilitates gefitinib-induced hepatotoxicity by degrading COX6A1 (cytochrome c oxidase subunit 6A1). Autophagy. 2021: 17(10): 3221-3237.
17] M. Platani, L. Trinkle-Mulcahy, M. Porter, A.A. Jeyaprakash, W.C. Earnshaw, Mio depletion links mTOR regulation to Aurora A and Plk1 activation at mitotic centrosomes, J. Cell Biol. 2015: 210: 45–62.
[18] Z. Li, X. Zhang, Kinases Involved in Both Autophagy and Mitosis, International journal of molecular sciences 2017: 18.
[19] T. Shao, X. Liu, Identification of Rictor as a Novel Substrate of Polo-like kinase 1, Cell Cycle.2015: 14: 755–760.
[20] TT. Liu, KX. Yang, J. Yu, et al. Co-targeting PLK1 and mTOR induces synergistic inhibitory effects against esophageal squamous cell carcinoma, J Mol Med. 2018: 96: 807–817.
[21] YF Tao, ZH Li, WW Du, et al. Inhibiting PLK1 induces autophagy of acute myeloid leukemia cells via mammalian target of rapamycin pathway dephosphorylation, Oncology Reports. 2017: 37: 1419–1429.
[22] S. Ruf, A.M, Heberle, M, Langelaar-Makkinje, S, et al. PLK1 (polo like kinase 1) inhibits MTOR complex 1 and promotes autophagy, Autophagy. 2017: 13: 486–505.
[23]Yata K, Lloyd J, Maslen S, Bleuyard J.Y, Skehel M, Smerdon S.J, Esashi F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol. Cell. 2012: 45: 371–383.
[24]Wang H, Qiu Z, Liu B, et al. PLK1 targets CtIP to promote microhomology-mediated end joining. Nucleic Acids Res. 2018: 46(20): 10724-10739.
[25]Pierce A.J, Johnson R.D, Thompson L.H, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999: 13: 2633–2638.
[26]Barton O, Naumann S.C, Diemer-Biehs R, Kunzel J, Steinlage M, Conrad S, Makharashvili N, Wang J, Feng L, Lopez B.S.et al. Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. J. Cell Biol. 2014: 206: 877–894.
[27]Steegmaier M, Hoffmann M, Baum A, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007: 17(4): 316-22.
[28]Hu K, Law JH, Fotovati A, Dunn SE. Small interfering RNA library screen identified polo-like kinase-1 (PLK1) as a potential therapeutic target for breast cancer that uniquely eliminates tumor-initiating cells. Breast Cancer Res. 2012: 14(1): R22.
[29]Bussey KJ, Bapat A, Linnehan C, et al. Targeting polo-like kinase 1, a regulator of p53, in the treatment of adrenocortical carcinoma. Clin Transl Med. 2016: 5(1): 1.
[30]Wissing MD, Mendonca J, Kortenhorst MS, et al. Targeting prostate cancer cell lines with polo-like kinase 1 inhibitors as a single agent and in combination with histone deacetylase inhibitors. FASEB J. 2013: 27(10): 4279-4293.
[31]Lu B, Mahmud H, Maass AH, et al. The Plk1 inhibitor BI 2536 temporarily arrests primary cardiac fibroblasts in mitosis and generates aneuploidy in vitro. PLoS One. 2010: 5(9): e12963.
[32]Rudolph D, Steegmaier M, Hoffmann M, et al. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res. 2009: 15(9): 3094-102.
[33]Brassesco MS, Pezuk JA, Morales AG, et al. In vitro targeting of Polo-like kinase 1 in bladder carcinoma: comparative effects of four potent inhibitors. Cancer Biol Ther. 2013: 14: 648–57.
[34]Rudolph D, Steegmaier M, Hoffmann M, et al. BI 6727, A Polo-like Kinase Inhibitor with Improved Pharmacokinetic Profile and Broad Antitumor Activity. Clin Cancer Res. 2009: 15: 3094–102.
[35]Gutteridge RE, Ndiaye MA, Liu X, et al. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol Cancer Ther. 2016: 15(7): 1427-1435.
[36]H. Döhner, M. Lübbert, W. Fiedler, et al. phase 2 trial of low-dose cytarabine with or without volasertib in AML patients not suitable for induction therapy, Blood. 2014: 124: 1426–1433.
[37]Palmer AC, Sorger PK. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell. 2017: 171(7): 1678-1691.
[38] Solanes-Casado S, Cebrián A, Rodríguez-Remírez M, et al. Overcoming PLK1 inhibitor resistance by targeting mevalonate pathway to impair AXL-TWIST axis in colorectal cancer. Biomed Pharmacother. 2021: 144:112347.
[39]Stehle A., Hugle M., Fulda S. Eribulin synergizes with Polo-like kinase 1 inhibitors to induce apoptosis in rhabdomyosarcoma. Cancer Lett. 2015: 365(1): 37–46.
[40]Gheghiani L., Shang S., Fu Z. Targeting the PLK1-FOXO1 pathway as a novel therapeutic approach for treating advanced prostate cancer. Sci. Rep. 2020: 10(1): 12327.
[41]Maire V., Nemati F., Richardson M., Vincent-Salomon A., Tesson B., Rigaill G., et al. Polo-like kinase 1: a potential therapeutic option in combination with conventional chemotherapy for the management of patients with triple-negative breast cancer. Cancer Res. 2013: 73(2): 813–823.
[42]Lian G, Li L, Shi Y, et al. BI2536, a potent and selective inhibitor of polo-like kinase 1, in combination with cisplatin exerts synergistic effects on gastric cancer cells. Int J Oncol. 2018 : 52(3): 804-814.
[43]Koncar R.F., Chu Z., Romick-Rosendale L.E., Wells S.I., Chan T.A., Qi X., et al. PLK1 inhibition enhances temozolomide efficacy in IDH1 mutant gliomas. Oncotarget. 2017: 8(9): 15827–15837.
公司介绍
