

EGFR(Epidermal Growth Factor Receptor)是上皮生长因子(EGF)细胞增殖和信号传导的受体。EGFR属于受体磷酸激酶(Receptor Tyrosine Kinases, RTK)HER 家族的四个成员之一,该家族包括EGFR (ErbB-1),HER2/ (ErbB-2),Her3(ErbB-3) 和Her4(ErbB-4)。研究表明,EGFR突变或过表达会引发肿瘤[1-2]。
人EGFR基因位于第7号染色体p13-q22区,全长200 kb,由28个外显子组成[3]。 EGFR是一种糖蛋白,属于酪氨酸激酶型受体,细胞膜贯通,分子量170KDa。EGFR广泛分布于哺乳动物上皮细胞、成纤维细胞、胶质细胞、角质细胞等细胞表面,EGFR信号通路对细胞的生长、增殖和分化等生理过程发挥重要的作用[4]。
EGFR的结构及生物学功能介绍


穿膜结构域(TM)是由23个氨基酸残基构成螺旋状结构的疏水区域,跨膜区锚定在细胞膜上。
胞内结构域(JM、TK、C端)是具有蛋白激酶结构域的细胞质内羧基端区域,共542个氨基酸残基,包含了3个子区域,分别是酪氨酸激酶区(TK),近膜区(JM)和C端末区(CTD)。酪氨酸激酶区有ATP结合位点,在EGFR与配体结合发生二聚化后,ATP与位点结合,激活下游信号通路。近膜区能够调节激酶二聚化,对下游信号通路有调节作用。C端末区在EGFR被激活时,发生自身磷酸化,磷酸化残基募集活化细胞内的信号转导途径[7-8]。
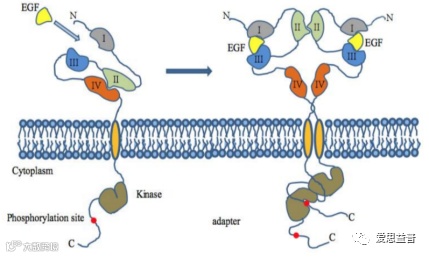
图1.3 EGFR通过EGF配体激活形成二聚体流程示意图[10]
EGFR受体可激活三个信号通路[11]:参与免疫调节的JAK/STAT信号通路,参与细胞增殖的RAS-RAF-MEK途径(MAPK/ERK通路),以及参与细胞存活的PI3K-AKT-mTOR途径(图1.4)。RAS-RAF-MEK途径负责控制基因转录活动和细胞循环周期,而PI3K-AKT-mT0R途径可激活抗细胞凋亡的信号。因此,EGFR受体蛋白在细胞增殖及存活上有着非常重要的作用。三种信号通路是细胞内信号转导的基础,调控肿瘤细胞诸多生理变化如:分裂、分化、生长以及迁移等。

EGFR过度表达,会激活下游信号通路,使得细胞生长无法抑制,肿瘤细胞增殖、转移等特性得以增强,最终促使肿瘤病变的发生[12]。
EGFR与肿瘤的关系和靶向药物研究进展
1980 年,研究者发现 EGFR 和 V-ERBB 之间高度同源,后者是一种病毒蛋白[13]。同时期从大鼠肿瘤中分离出属于 Erbb 基因家族的 Neu 促癌基因,发现在其跨膜结构域中存在单点突变[14]。这两份研究第一次发现了 EGFR 和癌症之间的关系。通过细胞和小鼠诱导 EGFR 过表达实验,证明 EGFR 可以促进癌细胞的增殖和转化并发展出相关肿瘤 [15] 。临床标本中也发现,在肺癌、食管癌和结直肠癌中存在 EGFR 基因过表达 [16] 。
EGFR与33%~50%的人类上皮肿瘤相关。乳腺癌、膀胱癌、肺癌及前列腺癌等许多恶性肿瘤中都发现有EGFR的过度表达,这说明EGFR在肿瘤细胞的恶性增殖中起重要作用。在大部分人类脑肿瘤中,EGFR基因都存在扩增或重排,由此产生的EGFR的过度表达在这些肿瘤的发生和发展中起重要作用 [17]。
通过对 EGFR 结构和调节机制研究了解,所研发的靶向 EGFR 的抗肿瘤药物可分为两大类,分别是单克隆抗体与小分子激酶抑制剂。
单克隆抗体包括西妥昔单抗(Cetuximab)、帕尼单抗(Panitumumab)、马妥株单抗(Matuzumab)等。这三种单克隆抗体的抗原表位都在 EGFR 的 III 结构域上,通过与EGFR胞外域的结合阻断配体与 EGFR 结合,从而阻止了下游通路的激活[18]。
小分子激酶抑制剂包括第一代EGFR-TKI靶向药物厄洛替尼、吉非替尼等,针对EGFR的黄金突变,即EGFR基因的19号外显子缺失突变(delE746-A750)和21号外显子的点突变(L858R)拥有良好的治疗效果。第二代EGFR-TKI靶向药物达克替尼、阿法替尼等,由于疗效较一代药物并未出现显著提升,且副作用更大,因此临床应用并不广泛。第三代EGFR-TKI靶向药物奥希替尼、艾维替尼等,已被作为EGFR敏感突变和T790M耐药突变NSCLC的治疗首选。他们是三磷酸腺苷(ATP)类似物,通过阻断胞内结构域的磷酸化来阻断信号通路的激活[18]。
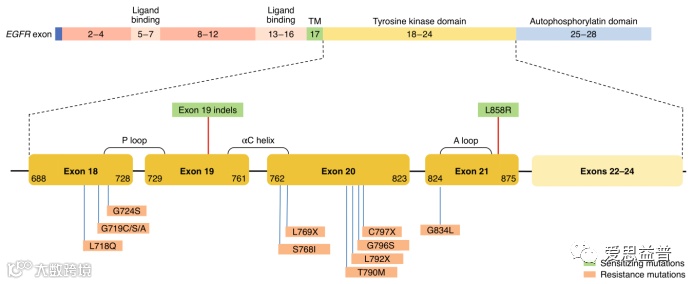
近十年来,以表皮生长因子受体酪氨酸激酶抑制剂(EGFR-TKI)为代表的分子靶向治疗,为癌症的治疗带来了巨大的变革,但EGFR-TKI的耐药性始终是一个悬而未决的难题。例如,EGFR-TKI在携带有EGFR突变的非小细胞肺癌(NSCLC)患者中一开始是有效的,但常演变为获得性耐药而失去治疗作用,如导致第一/二代EGFR-TKI耐药的T790M突变和导致第三代EGFR-TKI耐药的C797S突变等,尤其是同时含有L858R/T790M/C797S突变的肿瘤,目前已有的EGFR-TKI对其束手无策。
2.3 靶向 EGFR第四代新型变构抑制剂的研究进展

图2.2JBJ-04-125-02和JBJ-09-063的化学结构式[20]
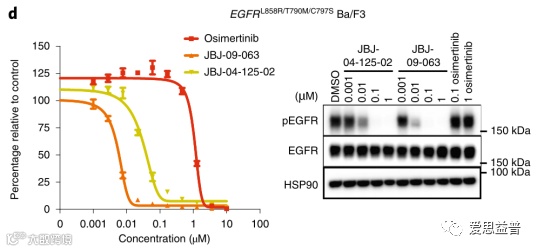
图2.2 JBJ-09-063比JBJ-04-125-02和奥希替尼抑制EGFR突变效果更好[20]
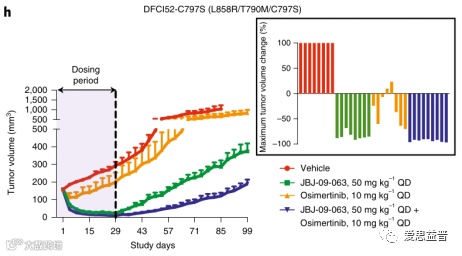
 图2.4 JBJ-09-063(a)和 奥希替尼(b)治疗不同突变的结合位点[20]
图2.4 JBJ-09-063(a)和 奥希替尼(b)治疗不同突变的结合位点[20]
图2.6JBJ-09-063可广泛有效地治疗对奥希替尼耐药的EGFR突变肿瘤[20]
JBJ-09-063可广泛有效地治疗EGFR-TKI耐药的EGFR突变肿瘤,然而在研究体外的H3255GR细胞 (细胞具有EGFRT790M突变,对吉非替尼耐药)时发现JBJ-09-063并不如在体内时那么敏感,而当ATP竞争性EGFR抑制剂吉非替尼(第一代EGFR-TKI)与JBJ-09-063联用后,可显著抑制H3255GR细胞的生长(图2.7),这说明JBJ-09-063和吉非替尼联用可逆转这一耐药性。

图2.7JBJ-09-063和吉非替尼 联用对EGFR T790M突变耐药性的影响[20]

2.4 靶向 EGFR第四代新型抑制剂BLU-945的研究进展

图2.9 化合物4、5、6化学结构式[25]
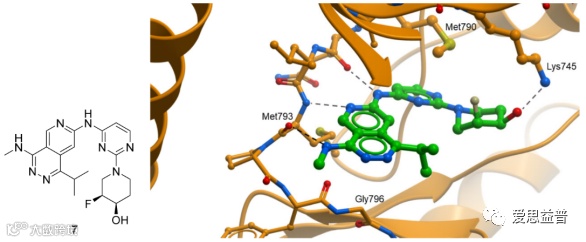
图2.10 化合物7和EGFRL858R/T790M蛋白X射线晶体结构解析图 (PDB: 8d73)[25]
通过SAR分析发现,将亚甲氨基换成环丁胺骨架时,化合物的活性及选择性有所提升。另外,在环丁胺的2位引入一个手性的甲基同样能够增强选择性及活性,不过,这会导致化合物具有较强的亲脂性,生物代谢稳定性不佳。在通过对环丁胺基骨架引入亲水性基团的筛选和尝试后,最终发现在3位引入一个含砜的结构 (化合物24,图2.11)可以有很好的活性与选择性,同时在体外的代谢实验中表现出很好的稳定性。
为了探究为何24的活性有所提高,研究者获得了24与L858R/T790M的EGFR蛋白的X射线单晶 (PDB: 8d76)。可以看到,砜上的其中一个O原子可以与Lys716和Lys728形成两个氢键,并进一步与邻近的α-helix中的Asp1003形成稳定的氢键网络。这种作用机制在已报道的EGFR抑制剂中并不常见,因此可能会是一个潜在的研究位点(图2.11)[25]。
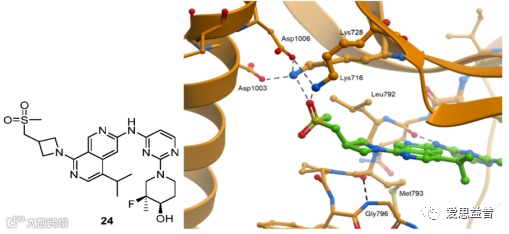
图2.11 化合物24和EGFRL858R/T790M蛋白X射线晶体结构解析图 (PDB: 8d76)[25]
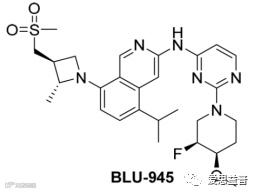
图2.12 化合物BLU-945化学结构式[25]

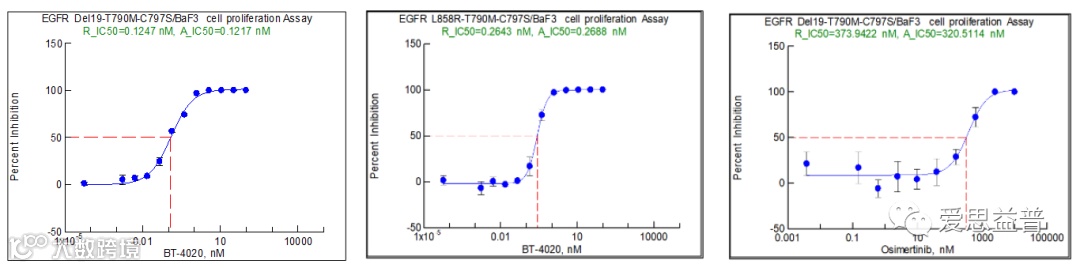
图2.13 不同浓度BLU-945和奥斯替尼对 BA/F3细胞系肿瘤模型的影响[25]
结语

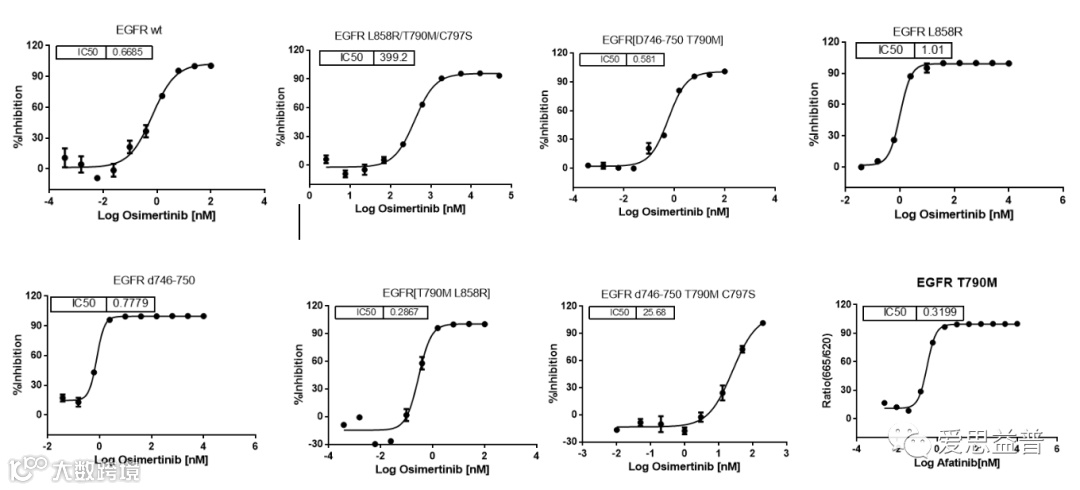
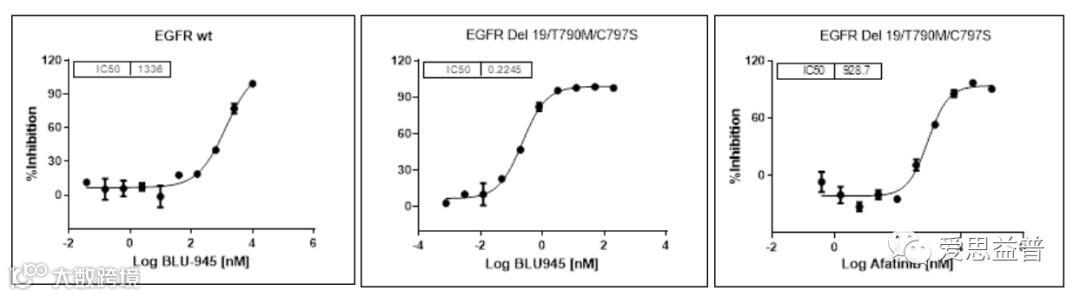
除了以上的研究方法,爱思益普还可通过HCS、Western Blot、qPCR、LC-MS等技术精确精准分析EGFR靶点、突变位点和不同细胞的耐药性。包括前期细胞培养、细胞生长曲线测定、目的化合物筛选分析等。ICE公司细胞种类贮备充足、设备前沿、理论知识充分,在实验中积累了大量的研发经验。为后期EGFR靶向药物的开发及优化奠定良好基础。
爱思益普以优质的服务和科技创新为根本,不断优化实验方法,不断科研创新,竭诚为国内外客户提供研发和检测服务,从而加速中国新药研发项目的进程。
参考文献
[1] Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer, 2005, 5(5),341-354.
[2] Scaltriti, M.; Baselga, J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin. Cancer Res, 2006,12(18), 5268-5272.
[3] International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature, 2004,431(7011), 931-945.
[4] Burgess, A.W. EGFR family: structure physiology signal ling and therapeutic targets. Growth Factors, 2008, 26(5), 263-274.
[5] SABBAH DA, HAJJO R, SWEIDAN K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors [J]. Cur Top Med Chem, 2020, 20(10):815-34.
[6] HERBST RS. Review of epidermal growth factor receptor biology [J]. Int J Radiat Oncol Biol Phys, 2004, 59(2 Suppl): 21-6.
[7] SHAO Q, ZHU WL. Ligand binding effects on the activation of the EGFR extracellular domain[J]. Chem Phys,2019,21(15) :8141-8151.
[8] Fukata Y, Murakami T, Yokoi N, et al. Local Palmitoylation Cycles and Specialized Membrane Domain Organization[J]. Current Topics in Membranes, 2016, 77(3),606-613.
[9] YARDEN Y. The EGFR family and its ligands in human cancer. Signalling mechanisms and therapeutic opportunities [J]. Eur J Cancer, 2001, 37 Suppl4(S3-8).
[10] Cheng Liang,Zhang Shaobo,Alexander Riley, et al. The landscape of EGFR pathways and personalized management of non-small-cell lung cancer[J]. Future oncology (London, England),2011,7(4).
[11] WANG L, ZHANG G, QIN L, et al. Anti-EGFR Binding Nanobody Delivery System to Improve the Diagnosis and Treatment of Solid Tumours [J]. Recent Pat Anticancer Drug Discov, 2020, 15(3): 200-11.
[12] Passaro Antonio,Jänne Pasi A.,Mok Tony,Peters Solange. Overcoming therapy resistance in EGFR-mutant lung cancer[J]. Nature Cancer,2021,2(4).
[13] Mo J S, Park H W, Guan K L. The Hippo signaling pathway in stem cell biology and
cancer[J]. EMBO reports, 2014, 15(6), 913-919.
[14] Yu F X, Guan K L. The Hippo pathway: regulators and regulations[J]. Cold Spring
Harbor Laboratory Press, 2013, 27(4), 336-349.
[15] Baselga J, Swain S M. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3[J]. Nature Reviews Cancer 2009, 9 (7), 463-475.
[16] Kleczko E K; Heasley L E. Mechanisms of rapid cancer cell reprogramming initiated by targeted receptor tyrosine kinase inhibitors and inherent therapeutic vulnerabilities[J]. Molecular Cancer 2018, 17 (1), 60-72.
[17] SIEGELIN M D,BORCZUK A C. Epidermal growth factor receptor mutations in lung adenocarcinoma[J].Lab Invest,2014,94(2):129-137.
[18] SPANGLER JB, NEIL JR, ABRAMOVITCH S, et al. Combination antibody treatment down-regulates epidermal growth factor receptor by inhibiting endosomal recycling [J]. Proc Natl Acad Sci U S A, 2010, 107(30): 13252-7.
[19] ROSKOSKI R, JR. ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors [J]. Pharmacol Res, 2014, 87(42-59).
[20] To Ciric,Beyett Tyler S,Jang Jaebong,Feng et al:An allosteric inhibitor against the therapy-resistant mutant forms of EGFR in non-small cell lung cancer. [J]. Nature cancer,2022,3(4).
[21] Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, Xu C, Rhee K, Chen T, Zhang H et al: Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric
inhibitors. Nature 2016, 534(7605):129-132.
[22] Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T et al: Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2018, 378(2):113-125.
[23] To C, Jang J, Chen T, Park E, Mushajiang M, De Clercq DJH, Xu M, Wang S, Cameron MD, Heppner DE et al: Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov 2019, 9(7):926-943.
[24] Jang, J. et al. Mutant-selective allosteric EGFR degraders are effective against a broad range of drug-resistant mutations. Angew. Chem. Int. Ed. (2020), 59,14481–14489.
[25] Eno Meredith S,Brubaker Jason D,Campbell John E,De Savi Chris,Guzi Timothy J,Williams Brett D,Wilson Douglas, et al. Discovery of BLU-945, a Reversible, Potent, and Wild-Type-Sparing Next-Generation EGFR Mutant Inhibitor for Treatment-Resistant Non-Small-Cell Lung Cancer.[J]. Journal of medicinal chemistry,2022,65(14).
公司介绍
