

从2023年8月EU GMP无菌附录的正式生效,到2025年初中国GMP无菌药品附录(征求意见稿)的发布,加上近期PDA Technical Report No. 22(TR22)《无菌灌装产品的工艺模拟》的正式发布,中国、美国、欧洲对无菌药品的“零污染”要求也完全保持一致了,中国对无菌生产企业APS的要求已经保持跟国际接轨,这对无菌制剂生产企业来说,面临着前所未有的合规挑战——监管机构对无菌制剂生产全流程的风险管控要求日益精细化,迫使企业重新审视现有APS与法规/指南要求之间的差距。无菌工艺模拟验证(APS)作为评估无菌生产过程控制能力的“金标准”,其科学性与严谨性直接关系到终端药品的安全底线。
PDA TR22最新版在延续经典验证逻辑的基础上,结合全球监管动态与技术创新,对模拟验证的风险评估框架、最差条件设定、数据科学应用等维度提出了更精细化的要求。
无菌工艺模拟是个复杂的知识点,也是大多数企业的薄弱点。我们将以新版TR22为核心框架,推出 「无菌工艺模拟验证实战解析」系列连载。本文将从对APS监管要求正确认识和APS基本概念和原则两点介绍。对于APS文件的要求、APS试验要素、干预设计和APS调查因素和风险评估工具的应用等要点将在后续文章中介绍。欢迎持续关注。
一、正确理解现在APS法规监管要求
未来的APS再也不是检查“有没有做”,而是“为什么这么做”,需要运用风险评估与科学分析方法为APS的设计决策提供重要依据,这些方法对于深化对无菌工艺及其能力的理解至关重要,最终需要通过风险评估呈现为什么这么设计。
欧盟GMP无菌附录中有一条的描述“9.32 无菌工艺中在线控制有效性的定期确认应包括使用无菌营养培养基和/或产品替代品的APS,APS不应被视为验证无菌工艺或无菌工艺方面的主要手段。”往往好多企业错误的认为APS就是或无菌工艺方面的主要手段。无菌工艺的有效性应当是一个成体系,科学性的工作。无菌工艺的有效性应当涵盖多方面的完整流程,至少包括:合理的APS工艺设计、设备确认、遵循完善的PQS、明确的工艺控制、过程监测与数据评估、人员培训,以及无菌工艺模拟(APS)。
二、无菌工艺模拟概念与原则
01
无菌工艺模拟的先决条件
对于新设施或生产工艺,APS作为整体验证活动的一部分实施。初始APS需要在完成以下环节后进行,我们可以理解成,当开始执行APS时,企业就已经完全具备商业化GMP的条件了,规避APS失败后的原因调查没有依据。
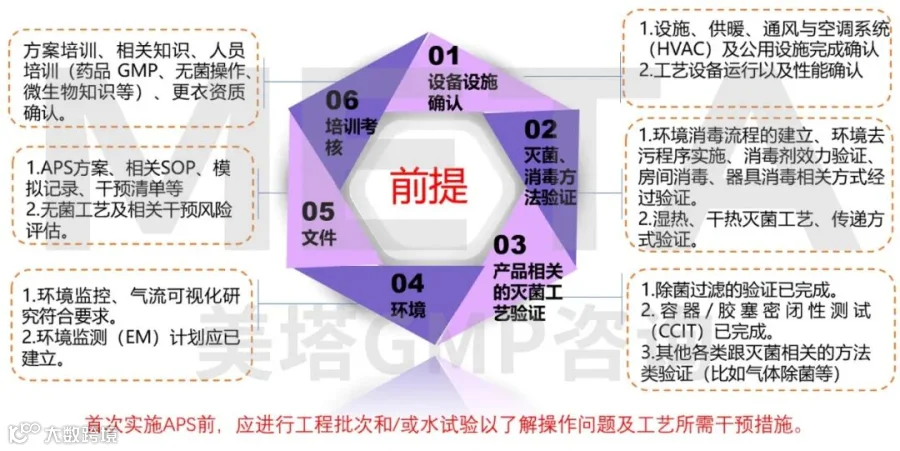
02
模拟次数与频率
以下汇总了中美欧标准的APS次数与频率的设计要求。现在的趋势是不管是首次模拟、还是周期性的模拟、还是事件驱动的再验证,均提倡使用基于对工艺带来的风险的评估决定,不再建议拍脑袋式的规定。

03
最差条件
APS中包含“最差条件”的目的是为了保证在常规工艺允许的极端情况下,尤其是对产品造成最大污染风险的情况下,生产无菌产品。例子包括最大无菌保持时间、最大允许在场人数等。最差条件不包括可能导致工艺偏差的情况,如设备故障、停电,或包括对工艺造成不可接受风险的干预。但往往这么设计了还是依然会被挑战到,拿灭菌后的器具存放时限举个例子,文件规定是72H,在模拟过程中到底是超过72H?还是接近72H但不超过?相信这两种方式可能均被挑战过。首先我们需要基于风险理解“工艺允许的极端情况下”,担心的是创造了允许之外的时限,导致APS的失败。而接近72H但不超过得形式确认可能非最差条件。我们在器具灭菌后存放有效验证需要验证时肯定需要验证超过72H,比如75H,只有75H都合格我们才能缩短控制到72H,基于前期的这个时限研究,72.5H可以被认定为是有保障的时限,基于风险评估输出72.5H,上述两种形式的风险均能被解决。
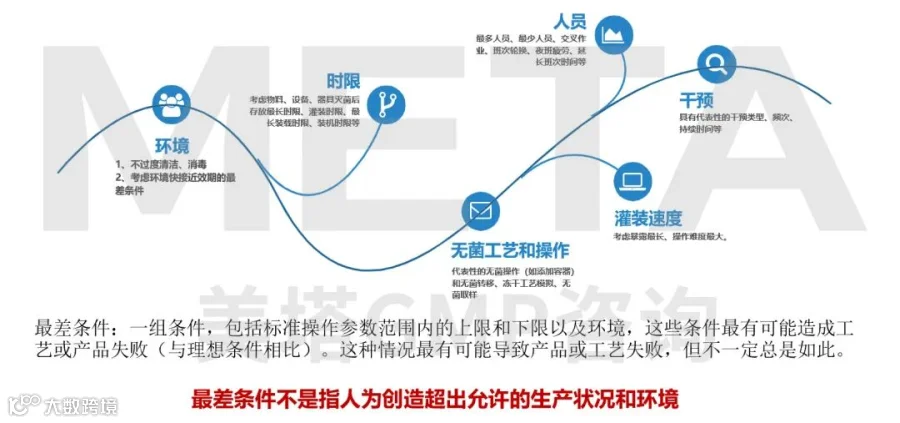
04
无菌工艺模拟设计风险评估
评估APS模拟的工艺流程,并为最终的APS研究设计提供依据。唯有在评估无菌工艺相关的危害与风险后,无菌工艺模拟设计方能被视为具有代表性且合理。应组建精通工艺、设备及设施设计的多功能团队,详细审查无菌工艺中污染物侵入的可能性,重点关注干预频率、干预操作与无菌物料及表面的距离,以及干预持续时间等作为无菌风险评估的关键因素。
PDA Technical Report No. 22(TR22)《无菌灌装产品的工艺模拟》中提出了问答方式,可以结合ICHQ9和问答的方式实现无菌工艺模拟设计风险评估。
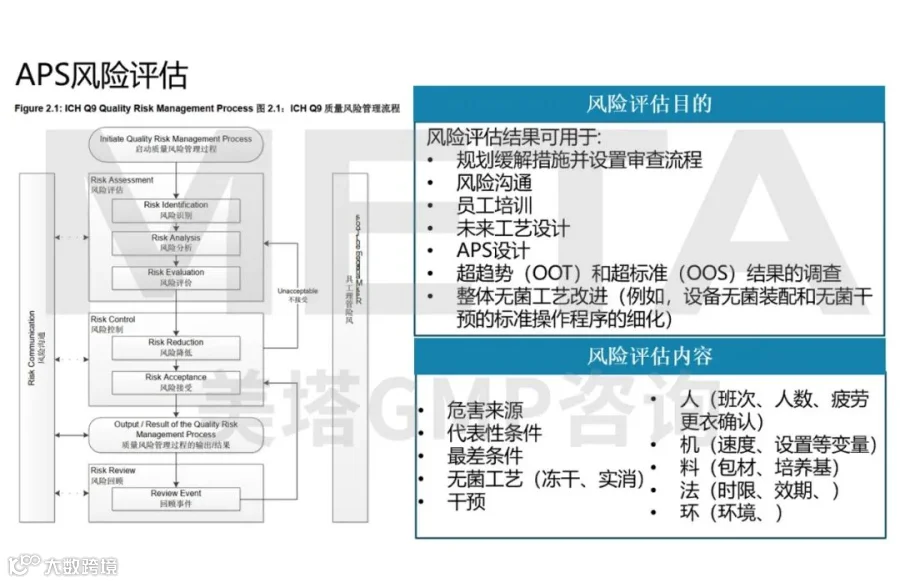
|
序号 |
问题描述 |
|
1 |
企业的无菌工艺系统设计是否充分涵盖生产全流程(包括评估/论证无菌工艺起始点),且未掩盖任何关键风险因素(例如:无菌工艺中使用惰性气体或冻干模拟过程中冷冻培养基可能影响污染物检测)? |
|
2 |
APS是否模拟了包括物料处理在内的整个制造过程? |
|
3 |
从一个子流程到另一个子流程的培养基灌装装置、操作设备和无菌干预的处理? |
|
4 |
企业的APS设计是否能够“挑战无菌过程的工艺能力”并识别以前未解决的变量? |
|
5 |
企业选择和模拟的最差条件制造场景是否与容器尺寸有关? |
|
6 |
企业配置、生产线速度、批量和操作条件是否合适,它们是否能够最好地代表无菌产品的风险,而不会给APS增加不必要的风险? |
|
7 |
企业的APS设计是否符合法规和行业指南要求? |
05
持续评估

参考文献
1. EU GMP Annex 1无菌药品生产
2. 中国GMP无菌药品附录(征求意见稿)
3. PDA TR22无菌灌装产品的工艺模拟(2025版)
4. NMPA 无菌工艺模拟试验指南(无菌制剂)
5. 中国GMP指南-无菌制剂(2023年版)
6. PDA Points to Consider No.1 Aseptic Processing (Revised 2023)
END
更多干货 请持续关注
【美塔GMP咨询】公众号
业务咨询

24小时内反馈
欢迎关注

微信公众号
关于美塔
美塔莫菲斯技术咨询(南京)有限公司是一家专注于为制药及健康产业提供全生命周期GxP合规解决方案的专业技术服务机构。我们以药品、医疗器械、化妆品、食品等领域为核心,通过“咨询+资源+落地”的一体化服务模式,助力企业构建符合NMPA、FDA、EU、WHO及PIC/S等国际标准的合规体系。



