


靶向甲酰肽受体1可减少脑部炎症和神经退行性变

「BioMedDaily」
研究概述


2025年11月13日,天津医科大学总医院施福东教授团队在Science在线发表题为“Targeting formyl peptide receptor 1 reduces brain inflammation and neurodegeneration”的研究论文。本研究揭示了FPR1在多发性硬化症进展中驱动持续性中枢炎症与神经退行性变的核心作用,并验证了FPR1拮抗剂的治疗潜力。

导读

「BioMedDaily」
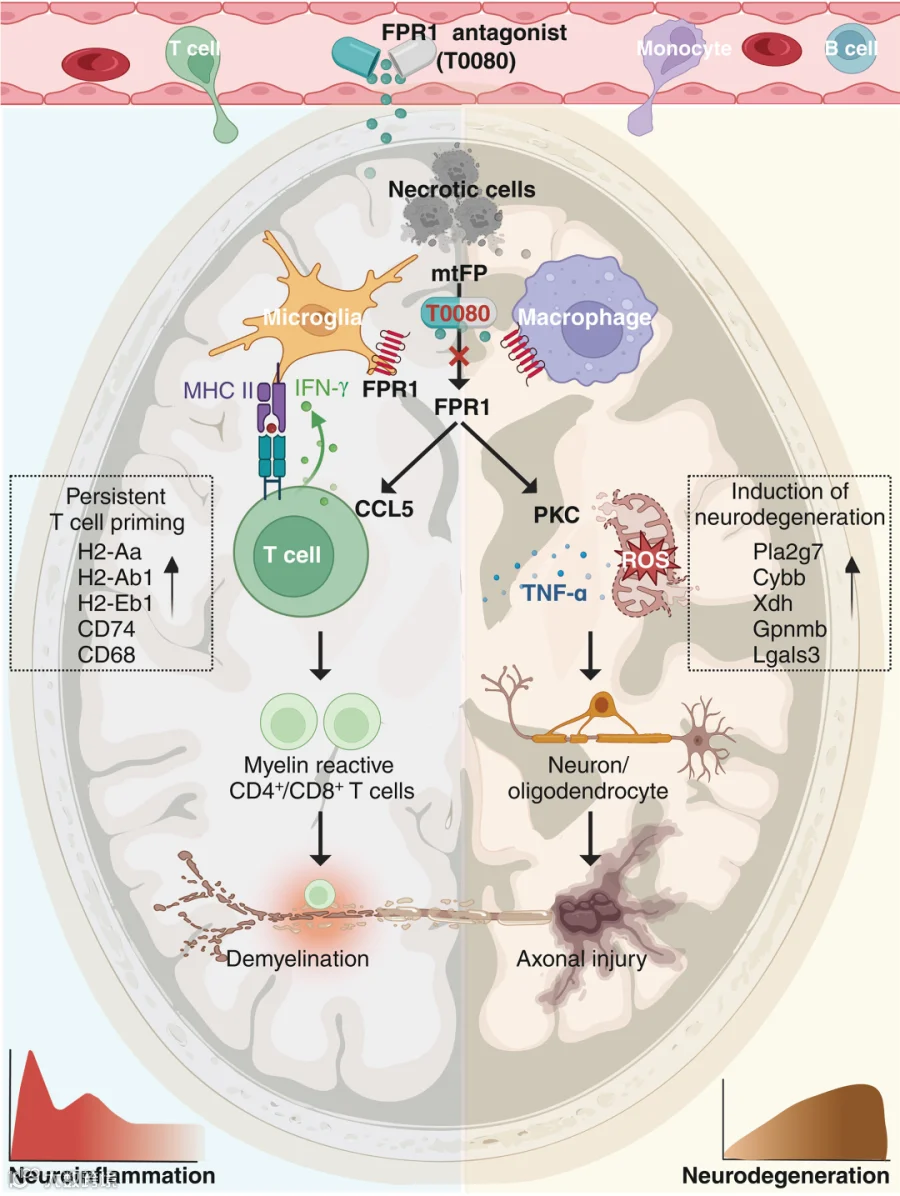
研究原理:活动期多发性硬化的病灶以小胶质细胞反应性活化为显著特征。反应性小胶质细胞与来自外周的浸润性巨噬细胞共同驱动病程中的局灶性炎症。持续的固有免疫反应会造成不断累积的组织损伤并加重神经退行性变。在疾病的慢性阶段,中枢死亡细胞释放危险相关分子模式,这些分子可被模式识别受体识别,例如主要表达于小胶质细胞和巨噬细胞的G蛋白偶联受体——甲酰肽受体1。甲酰肽与该受体结合后可触发强烈的固有免疫反应,加剧神经元损伤。此外,甲酰肽受体1的内源性激动剂——线粒体甲酰肽——由受损的线粒体释放,是神经退行性变的重要标志。
结果:研究团队发现,在多发性硬化患者的活动性脱髓鞘病灶中,甲酰肽受体1在中枢驻留小胶质细胞及外周单核细胞来源的浸润性巨噬细胞中显著上调。同时,循环中来自受损线粒体的N-甲酰肽水平会随疾病阶段动态变化。在多种小鼠模型中,作者进一步证明,甲酰肽受体1的激活可启动依赖蛋白激酶C的信号通路,诱导小胶质细胞和巨噬细胞持续产生活性氧和肿瘤坏死因子α,形成氧化与炎症环境,导致轴突损伤和逐步解体。此外,表达甲酰肽受体1的小胶质细胞分泌趋化因子CCL5,建立促炎微环境,促进髓鞘反应性CD4阳性T细胞在中枢募集与扩增;这些T细胞分泌干扰素γ,形成正反馈环路,进一步加重髓鞘破坏并维持神经炎症。为评估靶向甲酰肽受体1的治疗潜力,研究团队使用可进入中枢的小分子拮抗剂T0080,结果显示其能够减轻外周和中枢的自身免疫反应,并显著抑制小胶质细胞驱动的轴突退行性变。
结论:研究表明,甲酰肽受体1信号在维持中枢神经炎症并加剧神经退行性变中起关键作用。该机制性发现表明,甲酰肽受体1及其小分子拮抗剂T0080具有成为减缓多发性硬化进展的新型治疗策略的潜力。
研究结果

「BioMedDaily」
研究团队在多发性硬化患者不同疾病阶段分析FPR1表达,并使用7T MRI观察脑病灶活动。结果显示,与健康个体相比,FPR1在活动性病灶中显著上调,主要分布于中枢驻留小胶质细胞及浸润性巨噬细胞,而在星形胶质细胞、神经元、树突状细胞、B细胞和T细胞中表达较低或不可检测(图1A、1B)。
通过比较活动性病灶、非活动性病灶及类正常白质的单核转录组数据,研究团队进一步确认FPR1在继发进展型及复发-缓解型多发性硬化的活动性区域显著增加(图1C–H)。白质测序结果显示,FPR1主要表达于小胶质细胞及来自外周单核细胞的浸润性巨噬细胞,而非其他中枢驻留巨噬细胞亚群(图1F–H)。通路分析提示,表达FPR1的小胶质细胞/巨噬细胞富集于抗原呈递、趋化和炎性因子产生等功能通路,支持其在中枢炎症放大中的关键作用(图1I)。在脑脊液和外周血中,FPR1亦主要出现在来源于外周的巨噬细胞、单核细胞及中性粒细胞中。
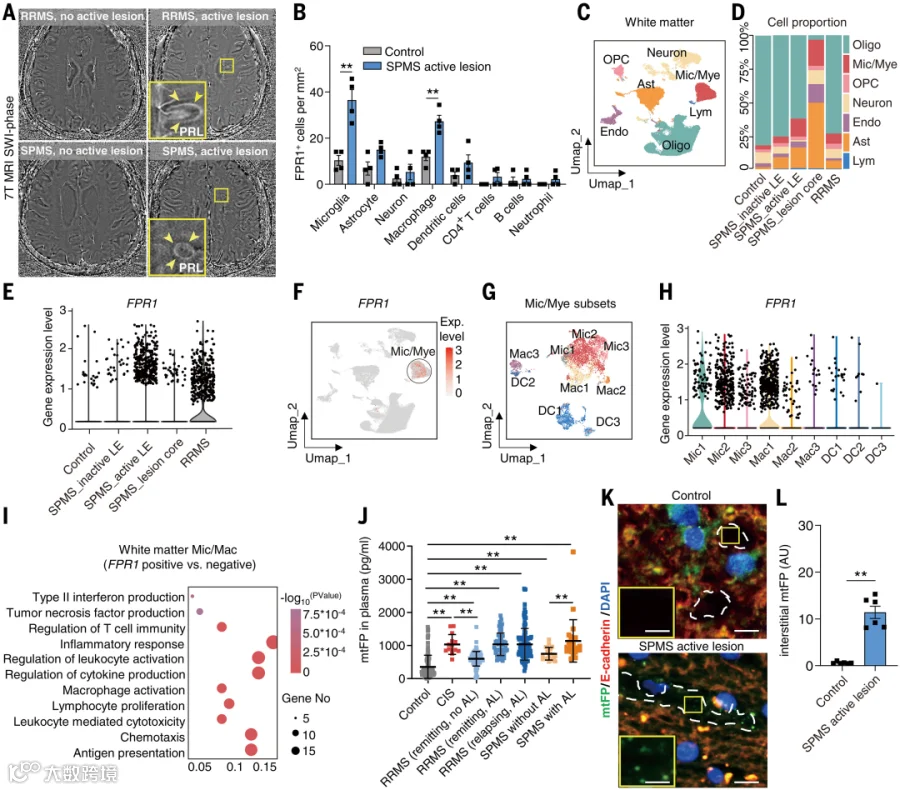
图1. 探究多发性硬化患者中甲酰肽受体1及其配体线粒体N-甲酰肽的作用。
研究团队进一步检测循环中线粒体甲酰肽这一内源性FPR1配体,发现其水平随疾病阶段升高,活动性病灶患者的血浆和病灶间质中尤为显著(图1J–L)。总体来看,活动性病灶中FPR1的显著上调及其配体的累积,提示FPR1信号通路在多发性硬化的中枢炎症与疾病进展中具有关键作用。
2. FPR1阳性的小胶质细胞和巨噬细胞会诱导脱髓鞘和轴突损伤
研究团队使用 MOG₃₅–₅₅ 免疫建立EAE模型,以评估FPR1在多发性硬化中的作用。在EAE高峰期,脱髓鞘白质间质、脑脊液和血清中的线粒体甲酰肽升高(图2A),伴随神经元与胶质细胞坏死,提示受损白质可释放 FPR1 配体。EAE小鼠中,FPR1 在小胶质细胞(CD45ⁱⁿᵗCD11b⁺)、巨噬细胞(CD45ʰⁱᵍʰCD11b⁺F4/80⁺)、中性粒细胞(CD45ʰⁱᵍʰCD11b⁺Ly6G⁺)和星形胶质细胞(GFAP⁺)中高表达,而在CD4⁺、CD8⁺ T 细胞及B细胞中表达极低(图2C、2D)。
为验证FPR1的致病作用,研究团队在小胶质细胞/巨噬细胞中特异性敲除Fpr1(Fpr1ᶠˡ/ᶠˡ × Cx3cr1ᴰʳᵉ 或 AAV-Cx3cr1ᴰʳᵉ-shFpr1),并使用全身敲除鼠(Fpr1⁻/⁻)。Fpr1缺失可显著减少脱髓鞘和轴突丢失,并改善运动行为(图2E–K)。在Fpr1ᶠˡ/ᶠˡ × Cx3cr1ᴰʳᵉ小鼠中,中枢小胶质细胞及浸润的巨噬细胞、T细胞数量下降(图2L),小胶质细胞中CD86、MHC II、TNF-α降低,IFN-γ⁺CD4⁺ T细胞比例减少(图2M),说明FPR1促进中枢炎症与组织损伤。

图2. 微胶质细胞/巨噬细胞中特异性敲除Fpr1揭示了FPR1介导病理在多发性硬化EAE模型中的关键作用。
为区分 FPR1 在中枢与外周免疫细胞中的作用,研究团队构建骨髓嵌合模型。删除中枢 Fpr1(WT BM→Fpr1⁻/⁻)可最显著改善脱髓鞘和神经缺损,而删除外周Fpr1(Fpr1⁻/⁻ BM→WT)仅有部分改善。综上,FPR1 在中枢小胶质细胞/巨噬细胞中的激活是驱动 EAE 脱髓鞘和神经退行性变的主要因素。
3. FPR1信号在小胶质细胞/巨噬细胞中呈现与神经退行性变和T细胞活化相关的特征
研究团队对WT与Fpr1⁻/⁻ EAE小鼠的中枢细胞进行单细胞RNA测序(图3A、3B)。在WT EAE小鼠中,FPR1主要表达于Mic1和Mac1亚群(图3C)。与Fpr1⁻/⁻相比,WT小鼠的Mic1/Mac1富集于抗原呈递、T细胞分化、趋化因子产生、TNF信号及细胞互作等通路,并显著激活FPR1下游MAPK和PI3K信号,提示FPR1增强其免疫反应性(图3D、3E)。与低FPR1表达的Mic2/Mac2相比,Mic1/Mac1同样富集MAPK、TNF及T细胞活化相关通路。
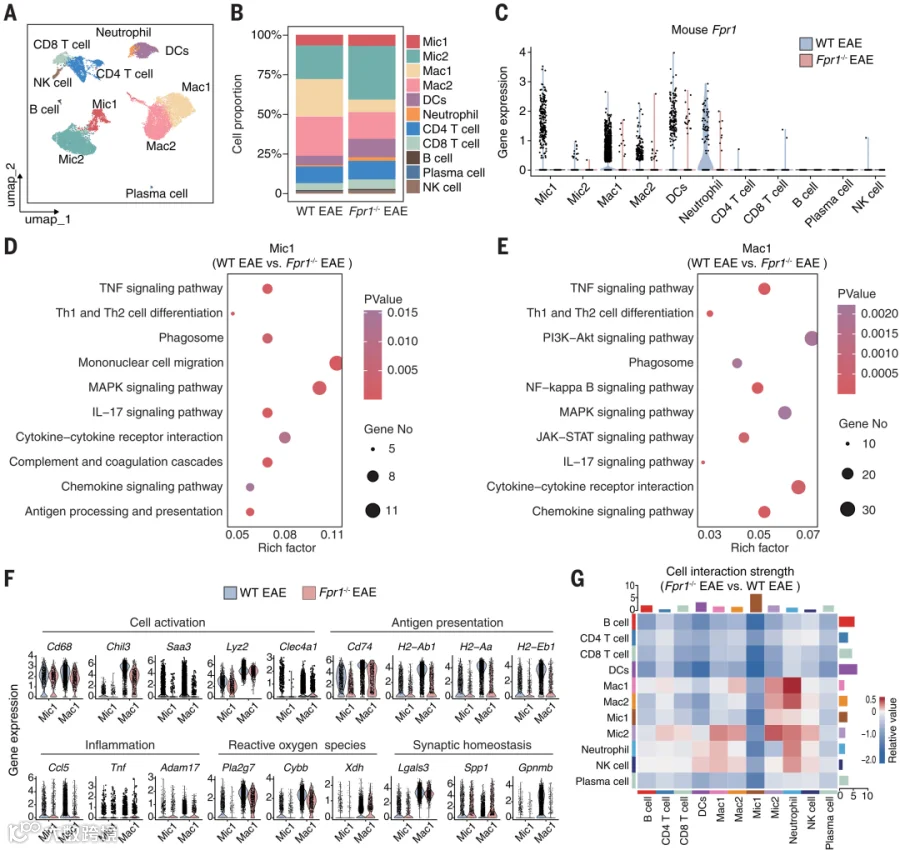
图3. FPR1决定中枢微胶质细胞/巨噬细胞的分子特征。
进一步分析显示,WT小鼠的Mic1/Mac1高表达细胞活化和代谢相关基因(Chil3、Saa3、Lyz2、Clec4a1)、炎症基因(Ccl5、Tnf、Amdm17)、ROS生成基因(Pla2g7、Cybb、Xdh)、抗原呈递基因(CD74、H2-Ab1、H2-Aa、H2-Eb1)和维持突触稳态的基因(Lgals3、Spp1、Gpnmb)(图3F)。这些变化未在Mic2/Mac2中出现,说明FPR1影响特定Mic1/Mac1亚群的基因表达。
此外,敲除Fpr1会削弱中枢细胞间的相互作用,尤其是Mic1与其他免疫细胞的互作(图3G)。总体来看,FPR1的激活可增强小胶质细胞/巨噬细胞的ROS和神经毒性因子产生,并促进其激活T细胞,从而推动EAE进展。
4. FPR1阳性的小胶质细胞维持髓鞘反应性CD4⁺ T细胞
EAE小鼠的脱髓鞘白质中,Fpr1缺失显著减少MHC II阳性小胶质细胞及其邻近的CD4⁺ T细胞(图4A、4B)。利用MOG₃₅–₅₅/I-Aᵇ四聚体分析显示,在病程高峰期WT小鼠的CNS内MOG₃₅–₅₅特异性CD4⁺ T细胞数量高于Fpr1⁻/⁻小鼠,而脾脏无差异(图4C、4D),提示FPR1促进CNS局部的致病T细胞扩增而不影响外周初始活化。
在CFSE标记的CD4⁺ T细胞共培养实验中,mtFP刺激可增强Fpr1⁺/⁺小胶质细胞/巨噬细胞促进T细胞增殖的能力,而Fpr1⁻/⁻细胞无此效应(图4E、4F)。在transwell体系中,激活FPR1可促进CD4⁺ T细胞迁移及其向IFN-γ阳性方向分化;抑制小胶质细胞的PKC或CCR5可降低CD4⁺ T细胞的迁移与IFN-γ表达(图4G–4I)。抑制IFN-γ进一步减少小胶质细胞MHC II表达(图4J),显示小胶质细胞与T细胞间形成促炎正反馈。
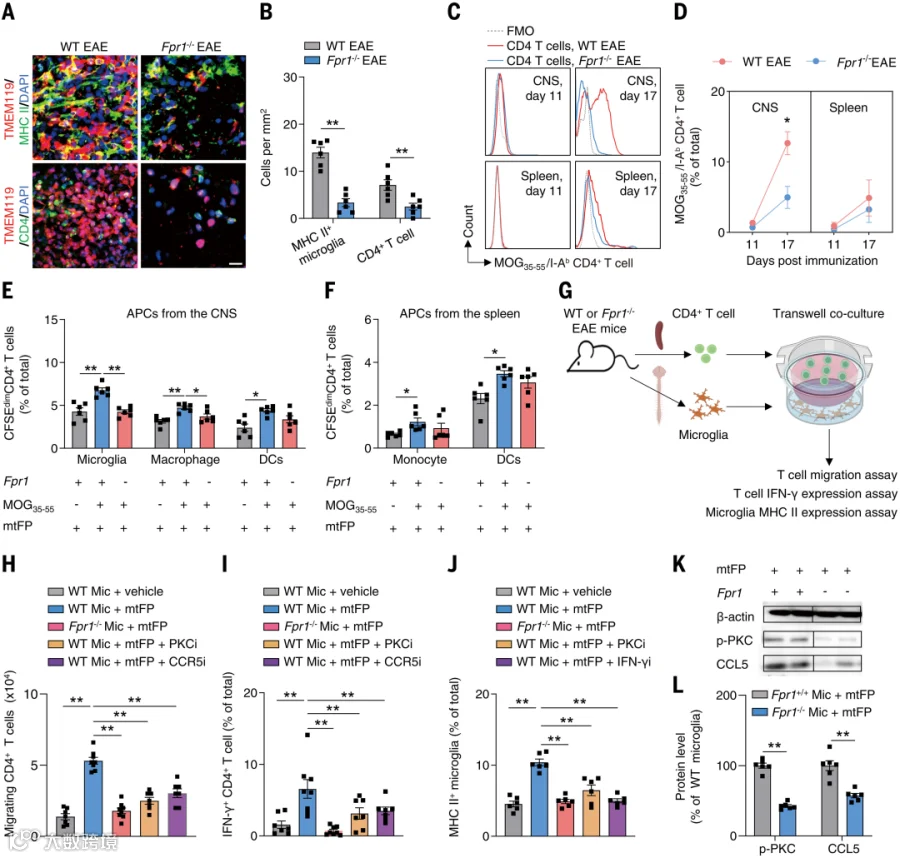
图4. 携带FPR1的小胶质细胞维持髓鞘反应性CD4⁺ T细胞。
单细胞测序和蛋白实验均表明,Fpr1缺失导致小胶质细胞/巨噬细胞的Ccl5和p-PKC水平下降,并降低CD4⁺ T细胞的IFN-γ表达,且Fpr1⁻/⁻小胶质细胞在mtFP刺激下产生的CCL5减少(图4K、4L)。此外,FPR1也促进CD8⁺ T细胞介导的神经损伤。综上,FPR1通过促进小胶质细胞释放CCL5、吸引并维持IFN-γ阳性CD4⁺ T细胞,从而增强CNS的抗原呈递与局灶炎症,推动EAE进展。
5. FPR1信号加速神经退行性变
单细胞测序显示,Fpr1缺失会降低EAE小鼠中枢小胶质细胞/巨噬细胞中与氧化磷酸化、ROS生成(Pla2g7、Xdh、Cybb)及突触稳态(Lgals3、Spp1、Fabp5)相关基因的表达(图3F)。WT EAE小鼠的小胶质细胞/巨噬细胞富集线粒体电子传递及ROS调控通路,而Fpr1⁻/⁻小鼠的NOX2明显下降,提示FPR1促进氧化应激。
在体外,小胶质细胞经mtFP激活FPR1后NOX2升高,而FPR1拮抗剂T0080可抑制其表达。体内发光成像显示,在EAE病程后期,Fpr1⁻/⁻小鼠脑内氧化应激显著降低(图5A、5B)。体外实验进一步表明,mtFP刺激可诱导小胶质细胞线粒体ROS增加及线粒体损伤,而T0080或NOX2抑制剂可明显改善(图5C–5E)。此外,FPR1激活可提升p-PKC、NOX2、LC3A/B及TNF-α蛋白水平,而Fpr1缺失可抑制这些变化并减少TNF-α、IL-1β分泌(图5F–5H),说明FPR1增强小胶质细胞的炎性与氧化反应。
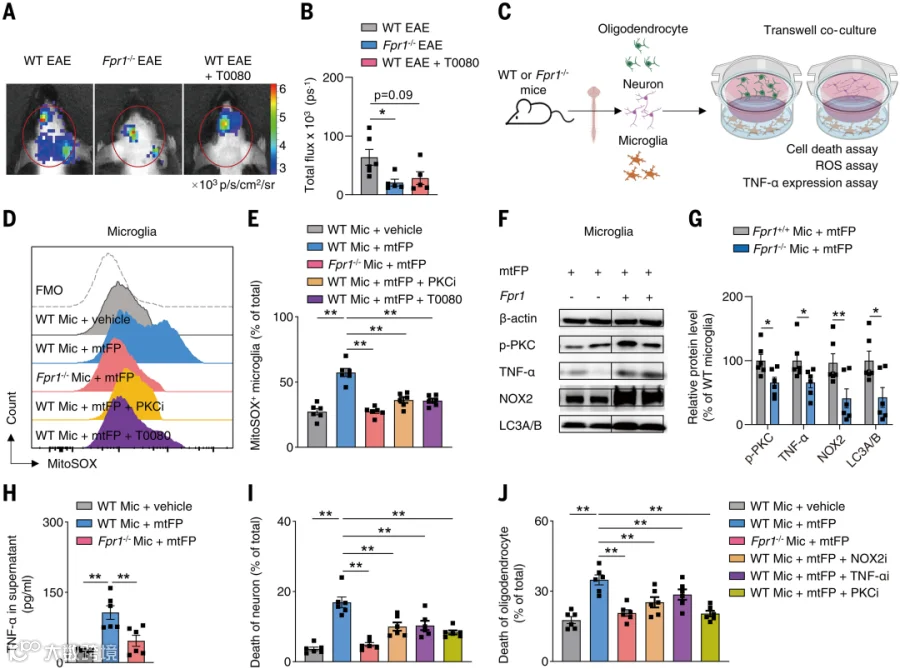
图5. 携带FPR1的小胶质细胞/巨噬细胞对轴突和少突胶质细胞具有损伤作用。
在共培养体系中,FPR1激活的小胶质细胞可导致神经元与少突胶质细胞死亡;Fpr1缺失或抑制NOX2、PKC、TNF-α均可阻断这一神经毒性效应(图5I、5J)。此外,FPR1活化可增加小胶质细胞产生CCL5,促进巨噬细胞向中枢募集,并增强其ROS与TNF-α释放,进一步加剧神经元和少突胶质细胞损伤。
在人源iPSC共培养体系中,FPR1激活同样可诱导神经元与少突胶质细胞凋亡;T0080或抑制PKC、NOX2、TNF-α均能有效保护细胞。综上,FPR1通过激活PKC并促进小胶质细胞/巨噬细胞释放ROS和TNF-α,从而诱导神经元与少突胶质细胞死亡,加速EAE模型中的神经退行性变。
6. FPR1拮抗剂T0080在三种MS小鼠模型中减缓疾病进展并改善病理
研究团队在三种MS模型中评估T0080的效应(图6A)。在MOG₃₅–₅₅诱导的C57BL/6 EAE模型中,T0080可显著降低神经功能缺损、脱髓鞘及中枢浸润的免疫细胞数量(图6B–6F),并减少脊髓的¹⁸F-FDG摄取,提示局部炎症下降(图6G、6H)。
为增强临床转化潜力,研究团队构建了人源化FPR1转基因小鼠。T0080在此模型中同样降低了临床评分与脱髓鞘程度,并减少表达人体FPR1的小胶质细胞数量。在更类似进展型MS的NOD-EAE模型中,疾病进展期小胶质细胞与巨噬细胞的FPR1上调明显。T0080治疗可改善30–60天阶段的临床表现(图6I),并减少脱髓鞘与轴突损伤(图6J–6M)。
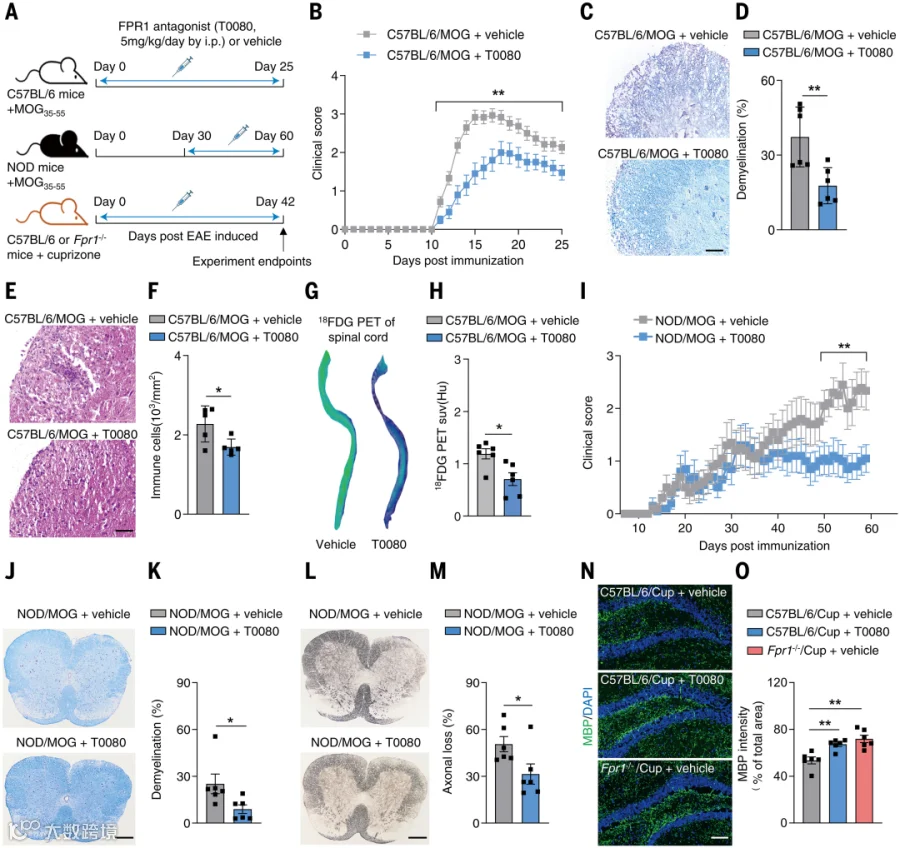
图6. FPR1拮抗剂T0080可在三种MS小鼠模型中减轻EAE病情。
在cuprizone脱髓鞘模型中,Fpr1敲除或T0080给药(5 mg/kg/天)均可减轻海马与胼胝体的髓鞘碱性蛋白丢失(图6N、6O),并伴随更少的小胶质细胞累积。综上,T0080在三种不同MS相关模型中均能抑制炎症反应、减少脱髓鞘与神经损伤,证明靶向FPR1具有广泛的治疗潜力,为其作为MS新型治疗策略奠定了实验基础(图6)。
讨论
反应性胶质增生是一个相互依赖的过程,会在MS进入进展期时维持中枢神经系统炎症并诱发神经退行性变。因此,以神经胶质细胞为靶点可能是阻止进行性神经退行性变的有效策略。既往研究显示,在cuprizone诱导的脱髓鞘模型中,FPR1在皮层上调且其缺失可减少脱髓鞘;此外,MS患者外周免疫细胞中也检测到FPR1升高。本研究进一步揭示,MS患者的小胶质细胞和浸润性巨噬细胞中FPR1显著上调,循环中内源性FPR1激动剂mtFP增加且其水平与MS病程相关,为探索FPR1在疾病进展中的作用提供了强有力的依据。删除小胶质细胞/巨噬细胞中的Fpr1可显著降低EAE小鼠的CNS炎症与神经退行性变。进一步研究发现,FPR1通过PKC信号增强小胶质细胞/巨噬细胞产生ROS和TNF-α,并降低其抗原呈递能力,从而共同推动神经炎症和神经损伤。
来自MS患者和EAE模型的证据表明,外周髓鞘反应性T细胞的产生、进入CNS以及克隆性扩增是疾病发起的关键步骤。在MS的慢性阶段,自身免疫反应逐渐局灶化于CNS。本研究结合原位表达、骨髓嵌合模型及单细胞测序表明,尽管外周免疫细胞也表达FPR1,但其致病作用主要局限于CNS内的小胶质细胞和巨噬细胞。研究团队在MS患者及EAE小鼠的活动性脑病灶中发现细胞坏死与间质mtFP,说明受损CNS组织可释放mtFP,通过刺激小胶质细胞和浸润免疫细胞维持免疫反应。
本研究提供了强有力的证据,表明FPR1信号在慢性MS病灶中介导神经退行性变。慢性活动性病灶的边缘区域富含表达MHC II或CD68的小胶质细胞/巨噬细胞,而在EAE模型中,FPR1敲除降低了这些标志物及抗原呈递相关分子的表达。进一步证明,FPR1通过与表达IFN-γ的CD4⁺ T细胞互作增强小胶质细胞的MHC II表达,促进其呈递抗原并驱动MOG₃₅–₅₅反应性CD4⁺ T细胞增殖,从而诱发髓鞘损伤。相比之下,由于树突状细胞和巨噬细胞表达FPR1较低或抗原呈递能力较强,FPR1对这些专业APCs的调节更为有限。FPR1⁺小胶质细胞是否与CD8⁺ T细胞或B细胞互作仍需进一步研究。
研究进一步揭示,FPR1信号通过激活PKC/NOX2通路引发线粒体损伤和线粒体自噬,促进ROS生成并诱导神经元凋亡。同时,FPR1激活促使小胶质细胞/巨噬细胞释放TNF-α,影响少突胶质细胞的存活,导致髓鞘丢失并阻碍再髓鞘化。因此,FPR1增强抗原呈递及ROS和TNF-α的产生,与慢性病灶中细胞构成、免疫特征及神经元/少突胶质细胞退行性变密切相关。
由于现有疾病修饰药物难以抑制CNS局部固有免疫反应和神经退行性变,它们对MS进展的改善有限。本研究确定了CNS内小胶质细胞与单核来源巨噬细胞上的FPR1作为潜在治疗靶点,其作用机制不同于现有MS药物。FPR1对CNS炎症和神经损伤的双重调控作用,以及其在CNS中的直接作用,使得开发如T0080的FPR1拮抗剂成为潜在有效的MS治疗策略。
你可能还想看
Nature | 注射到羊水中的人细胞会进入胎鼠器官,用于开发具有人类细胞的小鼠
Nat Commun | 减少CNS中过量的游离胆固醇可能是一种可行的ALS治疗策略
Nat Rev Drug Discov | 神经退行性疾病临床药物开发中的生物标志物指导决策
Nature | 在“100000基因组计划”中发现罕见病基因关联
Nat Rev Drug Discov | 罕见病患者个性化治疗方案:N-of-1疗法的发展与挑战
访问更多
Nat Med | 用于杜氏肌营养不良症的AAV mini-dystrophin基因疗法:一项1b期临床试验
Nat Biotechnol | 北京大学魏文胜团队合作报道基于Prime编辑器的高通量筛选揭示了人类细胞中的功能同义突变
Nat Commun | 天津医科大学郝继辉团队报道AI驱动优化Prime编辑器,扩展反向编辑窗口及增强保真度和效率
Nat Commun | 工程化U7小核RNA支架增加了ADAR介导的可编程RNA碱基编辑,在DMD中提高25倍
Nat Genet | 哈佛大学David R. Liu团队报道碱基编辑治疗三核苷酸重复序列导致的亨廷顿病和弗里德赖希共济失调
Nat Biomed Eng | Prime编辑结合多种递送策略治疗苯丙酮尿症
Nat Biotechnol | CRISPR–Cas13d:实现精准RNA靶向,破解非靶向降解难题
Nature | 杜氏肌营养不良症的基因补偿潜在疗法:转录适应
NEJM | 世界首次子宫内成功治疗遗传性罕见病:脊髓性肌肉萎缩症
Nat Rev Bioeng | 用于基因治疗递送的细胞外囊泡
Nat Biomed Eng | 长期植入人干细胞和祖细胞,用于在工程猪中大规模生产功能性免疫细胞
Nat Biotechnol | 通过靶向细胞表面RNA结合蛋白治疗急性髓系白血病
Cell Stem Cell | 利用人iPSC衍生的小胶质细胞在中枢神经系统范围内实现疾病修饰蛋白的精准递送
Nat Biotechnol | 浙江大学张进团队总结用于癌症免疫治疗的工程化先天免疫细胞
Nature | 靶向GD2的工程化改造细胞疗法治疗致命性儿童脑肿瘤
Nat Med | 基因修饰T细胞治疗后的肿瘤与病毒载体插入导致的突变无直接关联
Nature | 工程化心肌同种异体移植有效修复灵长类动物和人类心脏衰竭
Adv Drug Deliv Rev | 多能干细胞衍生细胞疗法治神经退行性疾病的临床观点
Nat Commun | 可扩展高质量的工程干细胞衍生的细胞外囊泡治疗急性肝衰竭
Nat Med | 异体间充质干细胞治疗阿尔茨海默:laromestrocel临床2a期试验
Nat Biotechnol | 非人灵长类动物中的脂质纳米颗粒筛选,递送效率高于小鼠,死亡率极低
Nat Nanotechnol | 通过PEG脂质比例和磷脂修饰调节脂质纳米颗粒的佐剂活性
Science | 脂质纳米颗粒设计用于在活体动物模型内产生治疗性T细胞,治疗癌症和自身免疫性疾病
Nat Commun | 用于快速将大分子治疗药物递送至外泌体的融合性脂质纳米颗粒
Nat Nanotechnol | 利用诱饵纳米颗粒减少肝脏吸收治疗性纳米颗粒,提高其肝外靶向
Nat Biotechnol | 用于核酸和蛋白质胞质递送的自组装蛋白质纳米颗粒,实现鼻内给药基因编辑
Nat Biotechnol | 使用脂质纳米颗粒实现更安全的非病毒DNA递送
Nat Mater | 用于中枢神经系统mRNA递送的跨血脑屏障脂质纳米颗粒
Small | 基于血脑屏障设计的纳米颗粒治疗脑缺血再灌注损伤
Nat Rev Bioeng | 用遗传工具探索细胞外囊泡的生命周期及药物递送潜力
「产业资讯」

罕见病基因治疗在进入临床试验后获得FDA批准的可能性为18.5%


转载须知
【原创文章】本文著作权归文章作者所有,转发分享请联系biomeddaily@163.com,未经作者的允许禁止转载,作者拥有所有法定权利,违者必究。

发现“赞”和“分享”了吗,戳我看看吧~
↓↓↓