
![]() 点击蓝字“代谢组metabolome”,轻松关注不迷路
点击蓝字“代谢组metabolome”,轻松关注不迷路

编译:微科盟 可乐,编辑:微科盟cordelia、江舜尧。
微科盟原创微文,欢迎转发转载。
小环肽作为一种治疗模式已获得广泛关注;然而,由于缺乏足够大规模的训练数据集,用于精确设计此类肽的深度学习方法发展缓慢。在此,本文提出一种名为AfCycDesign的深度学习方法,可实现环肽的精准结构预测、序列重设计及从头虚拟生成。借助AfCycDesign,本研究鉴定了超过10,000种结构多样性的设计产物,这些设计产物经预测能以高置信度折叠为目标结构。8个从头设计序列的X射线晶体结构与其设计模型高度吻合(RMSD<1.0Å),凸显了本方法的原子级精度。此外,研究以虚拟生成肽为初始骨架,设计出针对MDM2和Keap1的具有纳摩尔级别IC50的结合剂。本文开发的计算方法和骨架为针对不同蛋白质靶点及治疗应用的定制化肽设计奠定了基础。
论文ID
实验设计
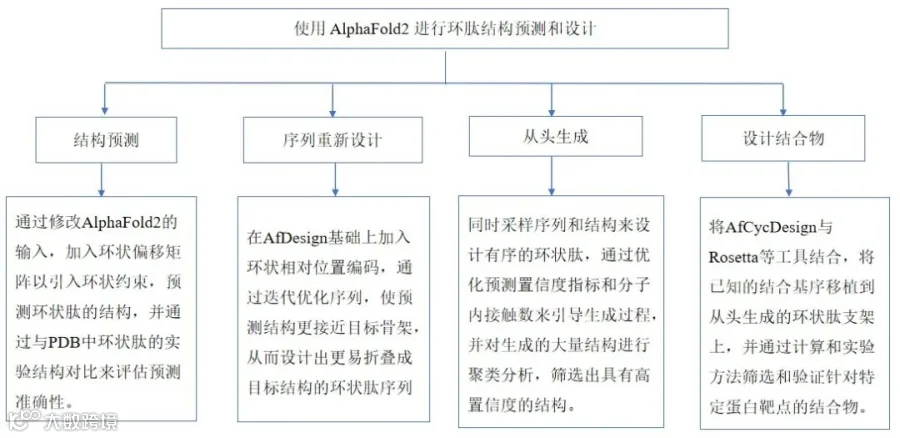
实验结果
1. 环肽的结构预测
本研究团队着手通过修改相对位置编码的输入参数来扩展AlphaFold2在环肽结构预测中的应用。对于线性肽,相对位置编码定义了残基间的序列间距——相邻残基的序列间距为1,而N端与C端之间的间距为肽链长度减1(图1a)。为了施加环化约束,研究者构建并应用了一个自定义的N×N循环偏移矩阵,该矩阵将环化信息引入相对位置编码,并将长度为N的肽链末端残基间的序列间距根据序列方向调整为1或-1(图1b)。经过独热编码和线性投影处理后,相对位置编码被添加到AlphaFold2网络中evoformer模块的成对特征中。如果缺乏此编码机制,注意力层将具有排列不变性和顺序不变性。本研究在ColabDesign框架中完成了上述改进,该框架基于AlphaFold2实现结构预测与设计,并将其命名为AfCycDesign。首先在蛋白质数据库(PDB)中随机选取的环肽序列上进行测试,发现初始测试输出的肽键连接和末端残基几何构型均正确无误,且未对肽链其他部分结构造成畸变。随后验证了输入序列为环化排列变体时,输出预测结果是否会发生变化。结果显示,所有环化排列序列的输出结构均表现出高度相似(图S1b)。
接下来,本研究评估了AfCycDesign对蛋白质数据库(PDB)中多种环肽结构预测的准确性。从PDB中收集了80个由标准氨基酸组成且序列长度小于40个残基的核磁共振(NMR)结构。这些结构未被纳入AlphaFold2的训练集,因为其训练排除了NMR结构及长度小于16个残基的短肽。这些结构涵盖了多种拓扑结构,且在尺寸、二级结构、序列及功能上具有多样性。值得注意的是,测试集中许多肽(如多种植物来源的环肽或环形结状折叠肽)含多个半胱氨酸残基和二硫键。二硫键连接方式的多样性(4个半胱氨酸有3种可能连接方式,6个半胱氨酸有15种可能连接方式)对此前基于Rosetta的方法构成挑战,且必须明确定义二硫键连接方式。使用AfCycDesign预测测试集中每个序列的结构,并评估两个指标:一是预测结构与实验测定结构的主链重原子均方根偏差(RMSD);二是预测局部距离差异测试(pLDDT),这是AfCycDesign输出的五个模型中用于评估结构预测置信度的指标。总体而言,AfCycDesign的预测结果与实验测定结构高度接近,pLDDT和RMSD的中位数分别为0.92和0.8 Å(图1b)。在80个测试用例中,有58个用例的预测结构显示出高置信度(pLDDT>0.7),且所有骨架原子相对于天然结构的RMSD小于1.5 Å。值得注意的是,在AfCycDesign 以更高置信度(pLDDT>0.85)预测的55个案例中,80%(n=44)的预测结构主链重原子与天然结构的RMSD小于1.5 Å,这表明pLDDT得分可用于筛选环肽的精准预测结果。此外,这些精准预测的结构不限于特定肽类或拓扑类型,涵盖了包括富含二硫键的环肽、小型环状β折叠和含极短α螺旋基序的肽在内的多种尺寸与拓扑结构(图1c)。在15个案例中,预测结构与实验结构非常接近(RMSD<1.5 Å),但AfCycDesign对这些预测的置信度较低(pLDDT< 0.85)(图1b)。
尽管未对二硫键连接施加额外的限制,但在大多数高置信度预测案例中,二硫键均形成了正确的连接方式,这对结状肽、芋螺肽、环肽等富含二硫键且序列众多但实验结构稀缺的肽类结构预测具有积极意义。相较于多序列比对(MSA),仅使用单序列进行预测在保证相近准确性的同时提高了预测速度——在单序列预测中,仍有49个预测满足pLDDT≥0.7且RMSD≤1.5Å(图1e)。相比之下,在单序列或基于MSA的预测中去除环化偏移会显著降低正确结构的预测能力(图1f)。上述RMSD值均基于最高pLDDT评分模型计算得出,此外,本研究还计算了天然结构NMR集合中所有构象与AfCycDesign五个预测模型间的RMSD(图S2)。在7个案例中,最高pLDDT模型未通过1.5Å的RMSD阈值,但其余4个备选模型中至少有一个预测结构的RMSD<1.5 Å。其中6例的最高pLDDT模型仍与NMR结构接近(RMSD<2 Å)。然而,在剩余1个案例(PDB ID:2B38)中,备选模型在RMSD方面的预测比 pLDDT 最高的模型显著提高。鉴于最高pLDDT模型并不总是最接近实验测定结构,本研究建议在后续任务中对AfCycDesign输出的所有5个模型进行评估。需要指出的是,即使仅基于单一模型进行预测,也显示出较高成功率(58/80正确预测用例),因此,研究人员决定将pLDDT作为本文所述研究任务的主要置信度指标。
本研究还探索了环状偏移的多种变体,以理解短程与长程相对位置编码以及编码方向性对环肽结构预测整体性能的影响。在偏移版本Type 1和Type 2中,根据偏移符号(+/-)对环序列中长程连接的方向性进行了不同处理。然而,两种偏移版本均显示出相似的结构预测精度。在AfCycDesign使用单序列与单循环设置预测的80个测试案例中,偏移Type 1和Type 2分别成功预测了52个和49个与天然结构接近且置信度较高(RMSD≤1.5 Å和pLDDT≥0.7)的肽。这些数据表明,序列空间中远距离残基的位置编码对结构预测精度无显著影响。为深入验证这一点,研究测试了Type3偏移——该版本仅提供相邻两个残基的相对位置编码,偏移矩阵中其他相对位置编码被设定为最大距离。研究结果发现,在相同的80个测试案例中,仍有48例能以良好置信度预测出接近天然结构的结果,说明仅通过相邻残基的相对位置编码施加环化偏移,便足以正确预测基准集中大部分环肽的结构。此外,研究人员还评估了肽的大小、二硫键存在与否或整体致密性是否会影响不同偏移类型的预测精度。结果显示,对于不同尺寸、不同紧凑程度或含有不同数量二硫键的环肽,使用Type 2或Type 3偏移时,预测准确性未出现显著差异。尽管这些偏移在预测基准集的结构时表现相似,但仍建议将Type 2作为默认偏移版本,因其包含的长距离位置编码信息有助于提升预测质量。
除预测准确性更高且无需明确指定二硫键连接方式外,与基于物理的方法(如Rosetta)相比,AfCycDesign需要的计算时间和资源要少得多,因为Rosetta需要大量列举结构-能量图谱。以基准集中某样本(PDB ID: 1JBL)为例,获取其结构-能量图谱需构建28,042个结构,并耗费120计算小时,耗时因肽链长度而异。相比之下,AfCycDesign仅需单个GPU运行2分钟即可准确预测该结构(补充图5b)。
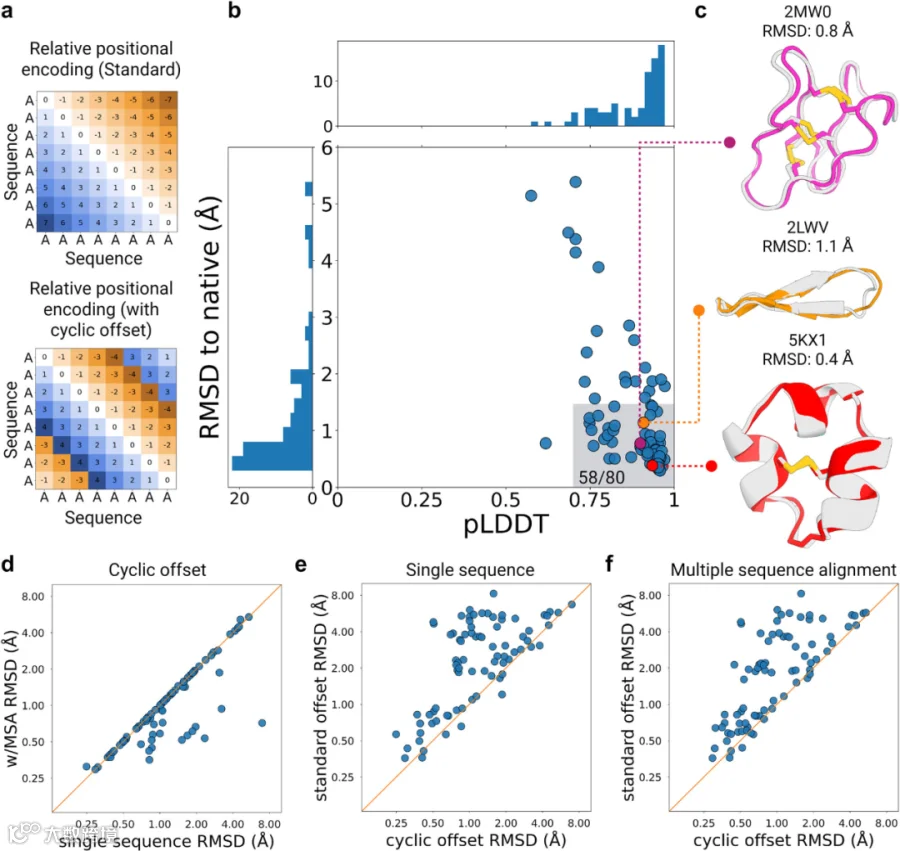
图1. 使用AfCycDesign对天然环肽进行结构预测。a.假设的八残基肽的相对位置编码示例。AfDesign中的标准编码显示线性肽残基位置间的序列间距,其末端彼此距离最远。AfCycDesign应用循环偏移改变了这一特性,使末端相互连接。b. AfCycDesign对蛋白质数据库中80个环肽的预测结果。高亮区域覆盖了高置信度且精确的预测(pLDDT>0.7,RMSD<1.5 Å)。c.三个代表性预测案例,展示了AfCycDesign以高置信度(pLDDT>0.85)正确预测(RMSD<1.5 Å)的多样化拓扑结构。实验测定结构显示为灰色,AfCycDesign预测结构分别显示为品红色、橙色和红色。d.AfCycDesign使用单序列循环偏移预测与使用多序列比对(MSA)预测的准确度(与天然结构的RMSD)对比。e.使用循环偏移与未使用循环偏移的单序列预测准确度对比。f.使用循环偏移与未使用循环偏移的MSA基础预测准确度对比。源数据详见源数据文件。使用BioRender创建。
2. 环肽的序列重设计
本研究进一步对AfCycDesign进行扩展,将环化相对位置编码应用于环肽骨架的氨基酸序列设计。推断该方法有助于识别能提升特定骨架折叠倾向性的氨基酸序列,这些骨架来源于天然存在的肽或通过其他骨架采样方法生成。为实现这一目标,研究人员在ColabDesign先前实现的AfDesign方法中引入了环化偏移。该方法的总体目标是通过AlphaFold2寻找能以高置信度折叠至目标骨架的预测序列。研究首先利用AlphaFold2网络从随机序列预测距离分布图(distogram),并在后续步骤中迭代优化序列,以最小化该步骤预测结构与目标骨架间的差异。序列优化以预测距离分布图(包含所有残基对距离的分箱分布张量)与目标结构中提取的距离分布图之间的差异(或分类交叉熵)为指导。此前研究表明,这种方式可有效最大化AlphaFold2的置信度,并最小化预测结构与目标结构之间的差异。
本研究致力于设计适用于靶向蛋白质-蛋白质界面常见螺旋-螺旋相互作用的肽骨架。研究人员使用Rosetta大环设计方法,生成了 457,615个13残基环肽的骨架,每个骨架均包含一个由7个氨基酸组成的短螺旋。为识别大规模筛选中所有独特的构型,本研究采用基于扭转角的分箱聚类方法对所得骨架进行聚类:每个氨基酸根据其ɸ、ψ和ω扭转角分配至特定箱,生成代表结构的箱字符串;其中分箱A和B对应拉氏图中的α和β区域,分箱X和Y对应图中ɸ角为正值区域的镜像区,同时,将分箱字符串的所有环化排列变体归为同一结构聚类。本研究鉴定出29,249个具有独特分箱字符串的聚类,并选择其中一个骨架RRR13.1(代表箱序列:AAAAAAXBYBBAB)进行重设计——因为针对该骨架的Rosetta设计序列在其能量图谱中能量间隔较小(ΔE < 2千卡/摩尔)(图2a)。AfCycDesign设计的序列与Rosetta设计序列差异显著,前者存在12处突变,仅核心区域的一个丙氨酸残基被保留。研究人员首先通过Rosetta环肽预测方法计算结构-能量图谱,在计算机中评估AfCycDesign生成序列RAR13.1的折叠倾向性。结果显示,AfCycDesign序列折叠为设计结构,且该结构为其最低能量构象;同时,设计结构与其他备选构象之间的能量间隔更大(ΔE ≈6.0 kcal/mol)(图2a、b)。为验证AlphaFold2设计序列是否确实折叠为设计结构,研究人员采用外消旋高分辨率X射线晶体学测定其三维结构,并与计算设计模型进行对比。结果显示,X射线晶体结构与设计模型高度吻合,Cα原子的RMSD为0.3 Å,且X射线晶体结构中13个侧链旋转异构体中有10个与设计模型匹配(图2c)。
鉴于AlphaFold2设计的RAR13.1结构验证成功,研究人员决定重新设计3274个独特结构聚类中的代表性肽进行重设计——这些聚类是从大规模骨架采样筛选中选取的,筛选标准为:骨架上搭载聚丙氨酸(或D -丙氨酸)序列时,Rosetta能量低于0 kcal/mol。同时,研究人员还使用Rosetta对所选骨架进行设计,并将其与AfCycDesign生成的序列进行比较。正如预期,对于相同骨架,AfCycDesign设计序列的pLDDT评分分布优于Rosetta设计序列(图2d)。Rosetta设计序列中仅有63个结构聚类的pLDDT>0.9,而AfCycDesign生成序列中有1,145个结构聚类满足pLDDT>0.9(图2d)。但需说明的是,Rosetta设计方法仅限于20种标准氨基酸(pLDDT计算所需),而无法如先前工作中实现的异手性设计。除比较结构预测置信度指标外,研究人员还探究了AfCycDesign与Rosetta设计序列在氨基酸组成及化学性质上的差异。结果显示,相较于Rosetta设计序列,AfCycDesign为相同骨架设计的序列通常更具疏水性,且含有更多脯氨酸(图2e)。总体而言,综合计算机模拟与实验结果证实,AfCycDesign可用于设计能折叠至目标结构的环肽骨架序列,且这些序列能折叠为目标结构。从更广泛的角度来看,AfCycDesign方法与其他肽骨架生成方法形成互补,二者结合可快速筛选出针对多种拓扑结构、且被预测能正确折叠的序列。
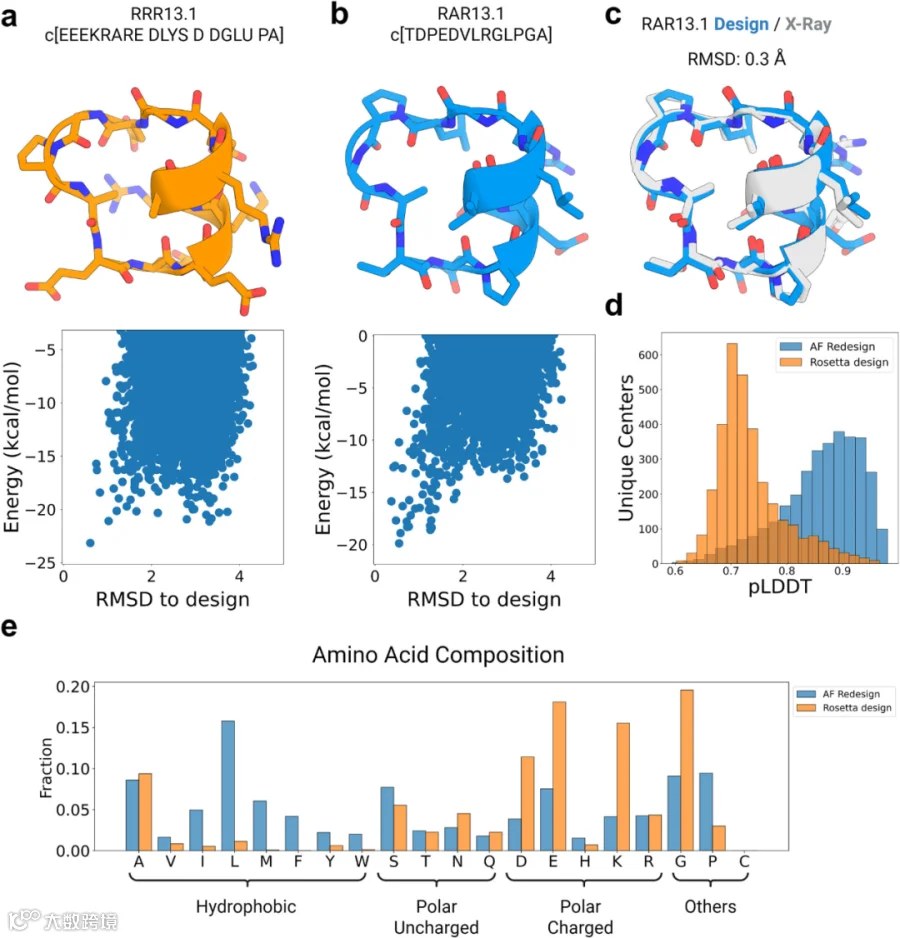
图2. 使用AfCycDesign设计环肽骨架序列。a. Rosetta设计的13残基环肽RRR13.1的序列与设计模型。序列中L-氨基酸用单字母代码表示,D-氨基酸用四字母代码表示。结构下方展示了Rosetta环肽预测方法计算的能量图谱(x轴为均方根偏差RMSD,单位Å;y轴为kcal/mol。b. AfCycDesign设计的13残基环肽RAR13.1的序列与设计模型。序列中L-氨基酸用单字母代码表示。结构下方展示了Rosetta计算的能量图谱。c. RAR13.1设计模型(蓝色)与高分辨率X射线晶体结构(灰色)的比对显示高度匹配,其Cα RMSD为0.3 Å。d. Rosetta(橙色)与AfCycDesign(蓝色)设计序列的预测局部距离差异测试(pLDDT)分数分布。各样本群均基于代表3274个独特13残基肽结构聚类的相同骨架设计。e. 在3274个独特13残基肽结构聚类骨架中,Rosetta(橙色)与AfCycDesign(蓝色)设计序列的氨基酸频率统计。源数据详见源数据文件。使用BioRender创建。
3. 环肽序列的重新生成
本研究进一步开发了一种生成方法,通过同步采样序列与结构来设计有序环肽,并将其应用于枚举结构多样的大环肽,而不仅仅是使用我们的重新设计方法生成的含螺旋13残基骨架的范围。该方法以损失函数为指导,这些损失函数旨在提高预测置信度指标(pLDDT和预测排列误差(PAE)以及分子内接触数量。
研究人员首先针对由7-10个残基组成的大环肽展开研究,为每种尺寸枚举48,000个生成模型。采用前述基于扭转箱的聚类方法对大规模采样结果进行聚类,分别鉴定出7残基、8残基、9残基和1残基环肽分别形成10,009、13,210、19,746和22,238个独特结构聚类(图3a)。在所有独特结构聚类中,7-10残基分别有200、296、486和1,352个聚类,其至少有一个成员会以非常高的置信度折叠成设计结构(pLDDT>0.9)。基于研究人员在天然结构预测与重设计中的结果(图1和图2),预期通过该严格置信阈值(0.9)的肽将正确折叠至设计构型。通过基于Rosetta的环肽结构预测方法这一正交技术,在计算机中对这些序列的折叠倾向性进行进一步验证。为评估这些序列的折叠倾向性,采用Rosetta环肽预测方法计算Pnear值:Pnear取值范围0-1,值为1表示设计结构是该序列唯一最低能量构象。大量生成序列在此计算中显示出良好折叠倾向,123个7残基肽、185个8残基肽、139个9残基肽和89个10残基肽序列的Rosetta Pnear值大于0.6。研究人员从7-10个残基的环肽中各选取一个满足AlphaFold2 pLDDT > 0.9且Rosetta Pnear > 0.9的生成设计模型进行实验验证与结构表征。所选的4个设计模型均缺乏规则二级结构,但通过大量分子内主链-主链及主链-侧链氢键作用保持稳定。RH7.1、RH8.1、RH9.1和RH10.1设计模型分别具有3个、5个、5个和6个分子内氢键(图3第二列)。所选模型的整体构型由经典α-转角、β-转角和γ-转角共同决定:设计模型RH7.1由I型β-转角与重叠的γ/α转角构成,所有转角均由脯氨酸残基成核;设计模型RH8.1包含两个I型β-转角,该转角通过天冬氨酸残基(i 位)与i+2位的亚氨基氢(NH)形成的侧链-主链氢键得以稳定;设计模型RH9.1同样含有两个I型β-转角,这两个转角通过甲硫氨酸-4与亮氨酸-9的长程氢键隔开,且该设计因仅含一个极性残基而呈现显著疏水性;设计模型RH10.1的序列同样高度疏水,含有多个暴露的非极性侧链,且色氨酸与亮氨酸之间通过疏水堆积作用稳定了分子内氢键作用较少的区域。此外,在所有设计模型中还观察到多个甘氨酸与脯氨酸,其中脯氨酸可提供构象约束,而甘氨酸可进入拉氏图中ɸ角>0度的X和Y分箱区域。
本研究通过化学方法合成了所选的4个肽,并采用高分辨率X射线晶体学测定其结构(图3第四列)。结果显示,RH7.1的X射线晶体结构与生成模型高度吻合,两者Cα均方根偏差(RMSD)为0.9 Å。设计模型与X射线晶体结构间仅存在微小差异:X射线晶体结构中,天冬氨酸侧链形成了一个额外的氢键,稳定了I型β-转角,而该氢键在设计模型中并未体现。RH8.1结构同样与生成模型接近(Cα RMSD 1.0 Å),但甘氨酸位点的ɸ扭转角与设计模型呈镜像翻转。RH9.1晶体结构与设计模型高度匹配(Cα RMSD 0.5 Å),RH10.1结构几乎与设计模型几乎完全相同(Cα RMSD 0.3 Å)。此外,RH10.1晶体结构中的侧链旋转异构体与设计模型也高度一致:两个亮氨酸和天冬氨酸旋转异构体完全相同,精氨酸旋转异构体的χ1、χ2和χ3二面角亦良好匹配,仅存在与天冬氨酸形成盐桥的微小偏差。
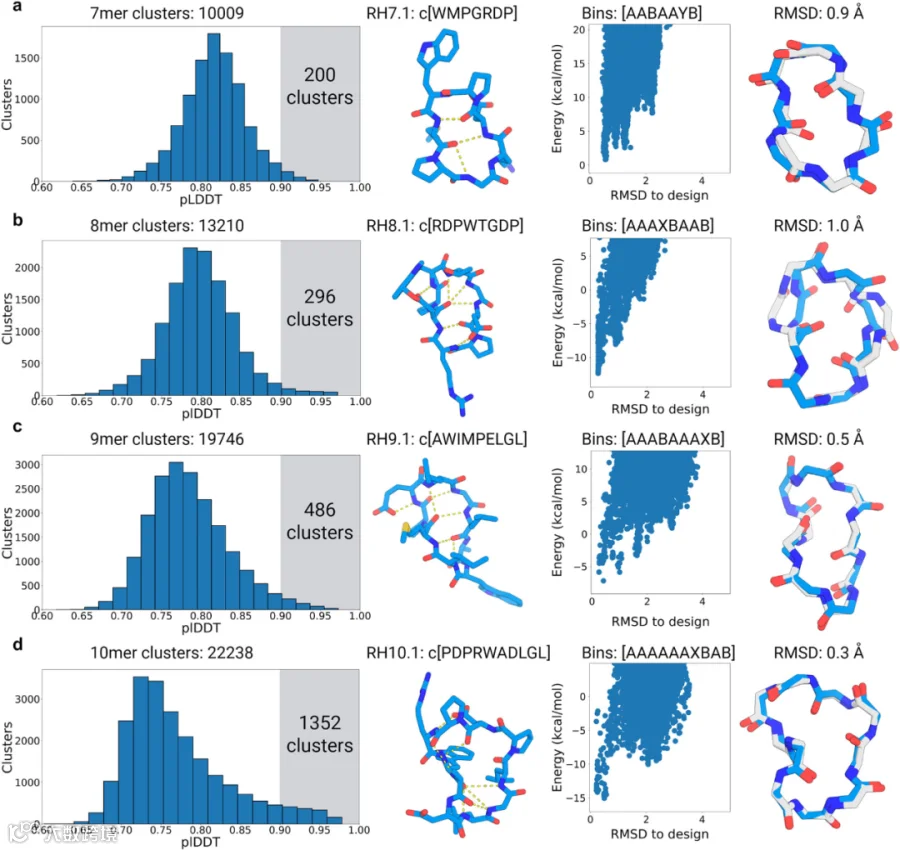
图3.使用AfCycDesign生成式设计7-10残基环肽。(a)7残基、(b)8残基、(c)9残基及(d)10残基大规模采样中预测局部距离差异测试(pLDDT)分布及候选序列验证结果。每行数据展示如下:第一列:7-10残基规模下48,000个生成肽段中识别出的所有独特结构簇的pLDDT分数分布,各规模独特聚类总数标注于图标题。图中高亮区域显示pLDDT分数>0.9的聚类数量。第二列:选定用于结构表征的生成模型结构及序列,氢键以黄色虚线标示。第三列:Rosetta计算的选定生成模型能量图谱(x轴为均方根偏差RMSD,单位Å;y轴为kcal/mol)。蓝色散点表示同一设计序列的不同构象(RH7.1样本量N=44,309;RH8.1 N=15,998;RH9.1 N=28,894;RH10.1 N=14,578),图谱顶部显示选定设计模型的扭转角分箱字符串。第四列:生成模型(蓝色)与X射线晶体结构(灰色)的比对结果。源数据详见源数据文件。使用BioRender创建。
随后,研究人员将生成拓展至11-13个残基大环肽设计。此前基于Rosetta的方法在较大环肽设计中面临显著挑战,需额外引入二硫键交联以稳定结构。为探究AfCycDesign是否能在无需额外交联条件下生成此类大环肽,通过大规模设计计算,分别鉴定出11残基、12残基和13残基大环肽的28,553个、27,64个3和27,330个独特结构聚类(图4第一列)。与7-10个残基的骨架不同(其独特骨架数量随残基数量增加而增加),11-13个残基独特聚类数量未随尺寸增加而稳定上升。这可能是由于仅对48,000个骨架进行采样不足以覆盖更大尺寸的环肽;若虚拟生成更多骨架,这些更大尺寸环肽的独特骨架数量可能会进一步增加。这也意味着,通过更大规模的虚拟生成筛选,仍有大量结构空间有待探索。研究人员发现有相当数量的聚类,其成员被预测能以高置信度折叠为设计结构::11-13残基分别有1,846个、2,86个1和3,825个聚类,其成员的AlphaFold2 pLDDT>0.9。从各尺寸选取一个满足pLDDT > 0.9且Rosetta Pnear > 0.9的序列进行实验验证。相较于较小环肽设计,11-13残基模型中出现了经典二级结构短基序(图4第二列):设计模型RH11.1含个8残基组成的α-螺旋基序,设计模型RH12.1和RH13.1则具有短延伸β-折叠。值得注意的是,12和13个残基长度对环状β-折叠而言并不典型(常见优选长度为6个、10个和14个残基的长度)。这些大环肽设计模型包含丰富分子内氢键(11-13残基分别含9个、7个和9个氢键)。RH11.1的短螺旋基序通过四残基延伸环完成环化,并由苏氨酸介导形成N端螺旋封端基序(图4a第二列)。RH12.1为短β-折叠,其一端通过经典II’型β-转角连接两条折叠链,另一端则为α-转角。RH13.1的链段配对因天冬氨酸侧链与主链酰胺氮形成的跨链氢键发生移位,形成由I型β-转角与α-转角两端环化的扭曲β折叠,并通过非极性侧链间的疏水作用进一步稳定(图4第二列)。
本研究通过固相化学肽合成法合成了RH11.1、RH12.1、RH13.1及其镜像异构体,并采用外消旋X射线晶体学测定三者结构。结果显示,所有三个肽的高分辨率晶体结构均与生成模型高度吻合,RH11.1、RH12.1和RH13.1的Cα RMSD 分别为 0.3 Å、0.4 Å和0.8 Å(图4第四列)。此外,X射线晶体结构中的转角类型与氢键模式均与所有三种肽的设计模型高度匹配。设计中的关键侧链相互作用在晶体结构中大多得以保留,最明显的两处偏差为:RH11.1中的色氨酸发生180°翻转(但仍保持与组氨酸形成环堆积作用,与设计模型一致);以及RH12.1中的酪氨酸不再与主链相互作用,转而与精氨酸形成阳离子-π相互作用。综上所述,这些数据充分证明了AfCycDesign在环肽从头生成中的出色准确性,包括无需依赖二硫键稳定的11-13个残基大环肽的生成,这一观点突破了既往理论认知。更广泛地说,本文所述的生成方法与大规模结构采样为功能整合提供了骨架。
接下来,研究人员通过计算位点饱和突变,并监测预测结构置信度指标的变化,评估了虚拟生成环肽的计算机模拟突变耐受性。针对含有7-13个残基的“高置信度”生成设计集合(n=10,681),对每个残基进行19种标准氨基酸替换并预测其结构。研究重点关注那些导致突变序列pLDDT显著下降(变化≥0.2)的突变类型。结果显示,这些骨架对大多数(98%)单点突变具有良好的耐受性,但对涉及脯氨酸和甘氨酸的突变除外:将甘氨酸突变为β-支链疏水性氨基酸(缬氨酸与异亮氨酸)时,分别有9.5%和10.8%的突变导致pLDDT下降≥0.2。最常见的pLDDT降低突变是亮氨酸突变为脯氨酸:此类突变中有16.1%导致pLDDT下降超过0.2个单位。意外发现亮氨酸突变为天冬氨酸时,9.8%的突变导致pLDDT下降0.2个单位。对此类设计的预测结构分析显示,天冬氨酸会破坏疏水簇。尽管这些短肽缺乏传统疏水核心,但设计的疏水区域/簇仍对其计算机折叠倾向性具有重要作用。
为比AfCycDesign生成的序列与ProteinMPNN设计的序列,研究人员还使用ProteinMPNN对22,182个10残基生成骨架进行序列设计,并比较两种方法的pLDDT表现。结果显示,两种方法设计的高置信度(pLDDT>0.9)序列数量相近:生成序列获得608个pLDDT > 0.9的序列,ProteinMPNN获得505个。然而,两种方法在不同类型骨架设计中各具优势,只有157个共同骨架被两种方法同时设计出高置信度序列(pLDDT > 0.9)。总体而言,这些数据表明,针对该环肽骨架集合,生成方法与ProteinMPNN的序列设计性能无显著差异。但由于二者可针对不同骨架实现高置信度设计,联合使用两种方法有望获得更多样化的高置信度设计。
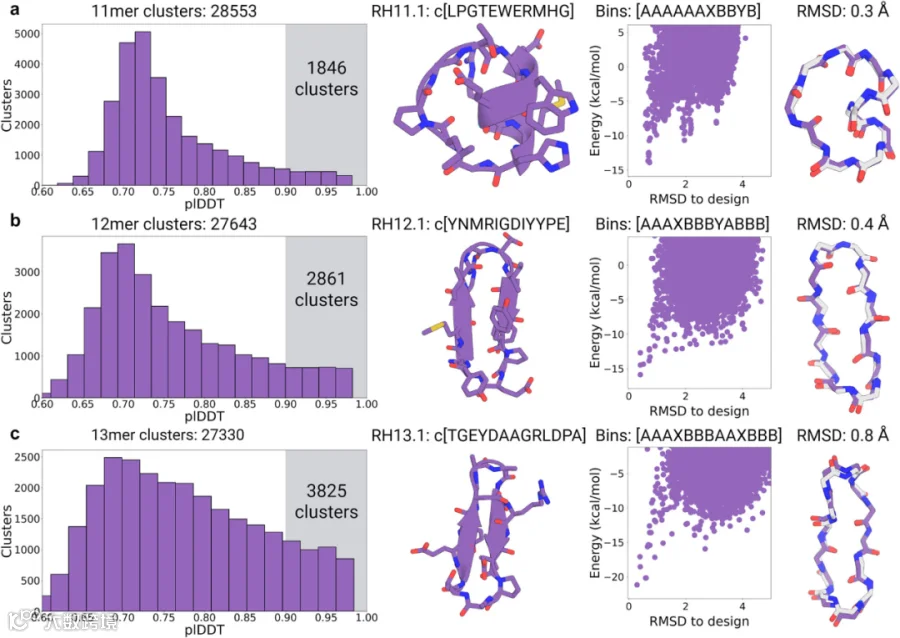
图4.使用AfCycDesign生成式设计11-13残基环肽。(a)11残基、(b)12残基及(c)13残基大规模采样中预测局部距离差异测试(pLDDT)分布及候选序列验证结果。每行数据展示如下:第一列:11-13残基规模下48,000个生成肽段中识别出的所有独特结构聚类的pLDDT分数分布,各规模独特聚类总数标注于图标题。图中高亮区域显示pLDDT分数>0.9的聚类数量。第二列:选定用于结构表征的生成模型结构及序列。第三列:Rosetta计算的选定生成模型能量图谱(x轴为均方根偏差RMSD,单位Å;y轴为kcal/mol)。紫色散点表示同一设计序列的不同构象(RH11.1样本量N=96,389;RH12.1 N=80,771;RH13.1 N=99,338),图谱顶部显示选定设计模型的扭转角分箱字符串。第四列:生成模型(紫色)与X射线晶体结构(灰色)的比对结果。源数据详见源数据文件。使用BioRender创建。
4. 基于生成骨架的环肽结合剂的设计与验证
接下来,本研究探索了AfCycDesign及生成环肽是否可用于设计特定蛋白靶点的结合剂。为此,本研究进一步改进了AfCycDesign,使其能够预测或设计蛋白-肽复合物,方法是仅对肽结合剂链施加环化偏移,而目标链保留默认位置编码。结论推断,这种预测蛋白-大环复合物的能力可用于基于预测置信度指标(pLDDT、界面预测对齐误差PAE等)筛选设计的大环结合剂,甚至实现结合肽的从头设计。在本研究中,研究人员探究了上述高置信度生成肽是否可作为初始骨架,通过在这些骨架上移植此前报道的结合基序和残基,设计针对蛋白质靶点的环肽结合剂。
作为原理验证,该研究首先选择设计靶向MDM2的结合剂,MDM2是一个已明确的治疗靶点,在肿瘤发生中具有多种作用,包括调节肿瘤抑制蛋白p53。结论推断,预先计算的高置信度虚拟生成肽(含短螺旋片段)是移植并模拟MDM2与p53之间天然螺旋介导相互作用的理想选择。在启动MDM2靶向设计前,研究将生成肽骨架的序列长度扩展至16个残基,获得24,104个经AfCycDesign高置信度预测(pLDDT > 0.9)能折叠至设计结构的多样化肽(图5a)。从p53(PDB ID: 4HFZ)中提取包含一个α-转角的5残基基序,该基序覆盖与MDM2抑制相关的色氨酸与苯丙氨酸关键残基。利用Rosetta软件套件中的MotifGraft工具,将该基序移植至生成骨架上(图5a)。移植后,通过三轮ProteinMPNN迭代设计及Rosetta能量函数优化,完成18,722个移植环肽的序列设计。根据现有文献明确色氨酸与苯丙氨酸对MDM2结合的关键作用,因此研究人员固定了该基序中这两个残基的序列,其余位点均进行重新设计。基于此前报道的ProteinMPNN在蛋白质结合剂设计中的成功应用,研究人员选择该工具进行序列设计。为在计算机中筛选设计候选物,使用AfCycDesign预测肽-靶标复合物结构。首先基于界面残基预测对齐误差(标准化iPAE < 0.3)对设计产物进行筛选,其中29%的设计产物通过该初筛。随后,进一步根据Rosetta界面质量物理指标(包括计算结合自由能ddG < -30 kcal/mol、空间聚集倾向评分SAP 评分< 30、接触分子表面积CMS > 300)及提高iPAE筛选阈值至<0.11,精选27个设计产物进行合成与测试。此外,由于11个设计产物的骨架残基(超出初始基序的部分)与界面区域的接触极少,或重新预测的结构偏离了原始基序位置,我们将这些设计产物排除在外。
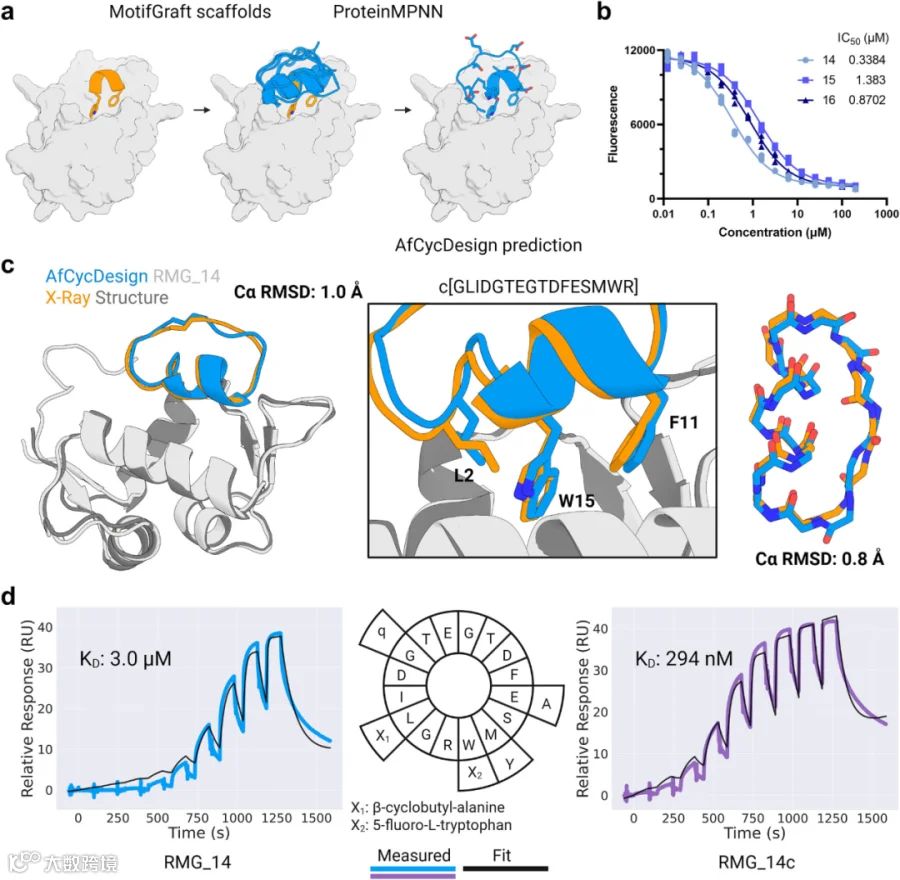
图5. 功能化生成式支架在体外抑制MDM2。a.将p53的五残基基序移植至7-16残基生成式支架组。移植支架序列通过三轮ProteinMPNN迭代设计及罗塞塔能量最小化获得。设计复合体由AfCycDesign基于单序列预测,并经过AfCycDesign置信度指标与罗塞塔评分体系筛选。b.三种最佳抑制剂在AlphaLisa检测中对MDM2的剂量效应曲线,半数抑制浓度(IC50)单位为微摩尔(µM)。各肽浓度荧光值呈三复孔检测。c. MDM2结合态RMG_14的X射线晶体结构与设计模型中MDM2的比对结果。肽链Cα RMSD为1.0 Å。中图显示设计模型与晶体结构关键界面残基构象一致。去除MDM2的肽链间比对显示设计肽段与晶体结构的Cα RMSD为0.8 Å。d. 表面等离子共振单循环动力学实验传感图:RMG_14与RMG_14c均采用9浓度点、3倍梯度稀释(起始浓度53.3 µM)。经5个氨基酸取代的变体RMG_14c(外环)显示出更高亲和力,解离常数(KD)分别为微摩尔(RMG_14)与纳摩尔(RMG_14c)量级。轮形图内环为RMG_14序列,外环标示RMG_14c突变位点,小写单字母代码代表D-氨基酸。源数据详见源数据文件。使用BioRender创建。
研究成功合成了16个选定环肽中的14个,且这些肽具有足够的产量和纯度,通过AlphaLISA实验对这些肽进行筛选,该测定法测量MDM2与先前描述的配体之间相互作用的破坏程度。在单浓度(50 μM)初筛中,14个设计产物中有5个显示出对MDM2-配体复合物形的抑制率超过50%。而作为对照的原始5残基基序在相同检测中未显示活性。随后选取在筛选实验中响应最佳的三个设计产物(RMG_14、RMG_15、RMG_16),并通过剂量效应AlphaLISA测定其半数抑制浓度(IC50)。结果显示,这3个设计产物的IC50值范围为0.34-1.38 μM(图5b),其中最优结合剂RMG_14的IC50达338.4 nM。这一结果凸显了实验流程能够识别蛋白质靶标的环肽结合物,即使对少量设计候选物进行低通量测试也是如此。
为验证RMG_14的结构及结合模式是否与设计一致,研究人员将MDM2结合的RMG_14复合物结晶化,并使用X射线晶体学方法确定其结构(图5c)。结果显示,1.7 Å分辨率的晶体结构与设计模型高度吻合:以MDM2为基准对齐时Cα RMSD为1.0 Å,单独比较肽段时Cα RMSD为0.8 Å。尽管基序对结合至关重要,但设计中其他残基在维持内部稳定性及与MDM2的相互作用中也发挥重要作用。
在初筛显示>50%活性的5个设计产物中,有2个(RMG_14和RMG_16)重现了p53中关键的FXXXWXXL基序,该基序对MDM2结合至关重要,并见于其他MDM2肽类结合剂。虽然两个疏水残基(FXXXW)属于移植基序且在序列设计中固定保留,但ProteinMPNN在无任何偏向性的情况下,仍在RMG_14的预期位置设计出亮氨酸。RMG_15及初筛中次优抑制剂RMG_13在该位点为甲硫氨酸,表明长链脂肪族残基在此位置具有耐受性。初筛中其他设计产物虽在此区域具有类似螺旋基序,但该位点为苯丙氨酸或异亮氨酸,这可能是其低活性或无活性的原因。然而,由于缺乏这些肽的实验结构数据,无法排除其未正确折叠或受溶解度、聚集等其他因素影响的可能性。
研究推断,生成环肽骨架在计算机模拟中表现出的高突变耐受性,因此可对已识别的活性肽进一步替换标准及非标准氨基酸,以提高其结合亲和力。在RMG_14中引入色氨酸类似物(5-氟色氨酸)、亮氨酸类似物(β-环丁基丙氨酸)及天然氨基酸替换(M7Y与E5A),因先前研究表明此类或类似替换可增强肽对MDM2的结合力。此外,研究发现RMG_14设计模型中存在一个ɸ角>0°的甘氨酸残基(G14),将其突变为D-型谷氨酰胺(D-Gln)可提升蛋白酶稳定性。通过G14→D-Gln,并进行其他4处替换,修改了近三分之一的原始序列。利用表面等离子共振技术,观察到这5个突变的引入使结合亲和力提升约10倍(图5d)。总之,这些发现表明,高置信度骨架集中的环肽骨架是肽结合剂设计的可优化初始模版。
受到MDM2结合肽的启发,本研究提出了一个假设,通过虚拟生成技术获得的肽骨架所具有的广泛结构多样性,能够容纳多种功能基序的移植,包括用于设计MDM2靶向大环的短螺旋以外的结构。为验证这一设想,研究人员尝试将1,014个先前被确认为介导环状结构参与蛋白质-蛋白质相互作用关键元件的“热环”,移植到建立的24,104个高置信度虚拟肽骨架上。研究结果显示,在这1,014个热环中,有798个能够成功移植到肽骨架上——每个热点环至少能与骨架集中一个环肽的4残基区域匹配,且Cα RMSD<1 Å和二面角RMSD<10°。完成基序移植后,可对骨架序列进行重设计,以与目标形成额外接触。虽然针对所有798个靶标进行结合剂设计与实验验证不具备现实可行性,但该研究选择以氧化应激和炎症应激反应的重要治疗靶点Keap1为模型,对所设计的结合剂进行实验表征。所有热环信息及其匹配骨架数据已完整收录于补充数据2中。
Keap1是一种依赖于Cul3的泛素连接酶复合体的受体,可识别并靶向转录激活因子Nrf2进行降解。研究人员采用了Gavenonis等人研究中报道的Nrf2蛋白DEETGE序列热点环,将其截短为4个残基的EETG基序用于移植,并筛选出775个与选定基序匹配的骨架(Cα RMSD<1 Å,二面体RMSD<10°)。接下来利用ProteinMPNN对骨架肽链的非移植区域进行序列重设计,并通过Rosetta软件进行基于能量的构象优化。在此过程中,研究人员又额外进行了3轮ProteinMPNN序列设计与能量最小化操作,每次输出一个序列,最终每个对接骨架得到4个序列。最后,采用AfCycDesign重新预测了大环结合Keap1的结构。基于ProteinMPNN输出的iPAE<0.15和RMSD <1.5 Å的严格筛选指标,研究人员选择6个设计产物进行实验验证,测试方法为竞争性荧光偏振(FP)检测(图6a)。所选的6个设计产物来自5个骨架,序列长度为12-13个残基。在这5个骨架中,1个具有明显的α-螺旋区域,其余则为类环或延伸构象。
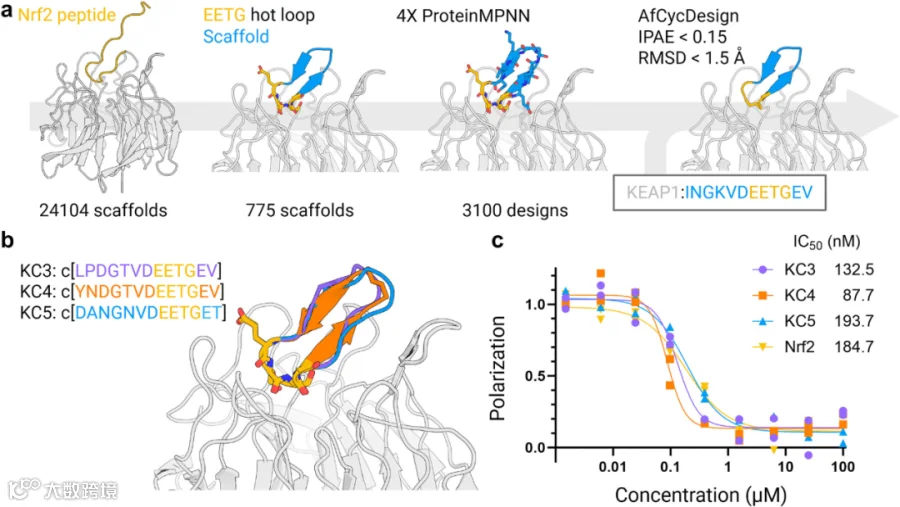
图6.通过将"热点环区"移植至生成式支架设计肽段结合剂。a.基于热点环区与生成式支架的结合剂设计流程。首图:16残基Nrf2肽段结合Keap1的结构(PDB:2FLU)。Nrf2的EETG区域被移植至AfCycDesign生成的24,104个高置信度支架。次图:775个支架与Nrf2热点环区匹配成功(均方根偏差RMSD<1Å,二面角均方根偏差<10°)。三图:所有775个支架经四轮ProteinMPNN迭代设计与罗塞塔能量最小化处理,每轮输出一条序列,热点环区残基EETG保持固定。四图:3100个设计经AfCycDesign预测,并通过界面预测对齐误差(iPAE)<0.15及预测肽段与设计模型RMSD<1.5Å的标准筛选。b.通过AfCycDesign指标验证的3个合成设计模型。c.竞争性荧光偏振检测(各浓度双重复实验)。晶体结构来源的16残基Nrf2肽段(PDB:2FLU)半数抑制浓度(IC50)单位为纳摩尔(nM)。源数据详见源数据文件。使用BioRender创建。
本研究成功合成了6个设计产物(KC1-KC6)中的3个,且其纯度和产量均符合实验要求,并将其与16个残基的Nrf2线性肽进行对比测试。实验结果显示,所有3个13残基的环状肽在结合活性上均达到或优于线性Nrf2肽(图6b)。其中KC4的活性是Nrf2的两倍,且无需进一步优化。尽管仅移植了4个残基的DEET基序,但所有3个设计产物均完整保留了原始热环基序DEETGE的结构特征。这三个设计产物均来源于不同的初始骨架,其中KC3和KC4的序列相似性最高(13个残基中有11个相同),主要差异在于KC4含有一个酪氨酸 ——该酪氨酸延伸至基序区域,并与基序中的D1形成氢键。这一相互作用可能进一步稳定了多肽的折叠构象,使其在FP实验中表现出优于KC3的活性。鉴于在虚拟单体结构验证及RMG_14-MDM2复合物结构预测中观察到的高精度,相信单独的设计模型可以用于进一步的结构引导优化,而无需耗时进行结构表征。KC4含有两个Φ扭转角为正值的甘氨酸残基,可以作为D-氨基酸取代的理想位点。综上所述,本文所建立的高置信度虚拟生成骨架集及相关方法,为开发靶向多种蛋白的大环化合物结合剂提供了坚实的理论与技术基础。
讨论
本研究提出一种在蛋白质结构预测网络中整合环状相对位置编码的方法,并利用其开发了多种关键应用的计算方法,包括环肽序列的结构预测、在天然及已设计的环肽骨架上重新设计氨基酸序列、针对不同拓扑结构的环肽进行从头虚拟生成,以及设计靶向治疗相关蛋白质的环肽结合剂。通过对蛋白质数据库(PDB)中已知环肽的结构进行测试,本方法展现出卓越的准确性:在80条环肽序列中,有58条预测结果与天然结构的RMSD≤1.5Å,pLDDT≥0.7,即预测结果准确;在高置信度预测结果中(pLDDT≥0.85),80%的预测结构与核磁共振测定结构的RMSD<1.5 Å。该方法的结构预测精度将为天然环肽提供快速可靠的结构解析支持,并对计算设计的环肽实现更精准的筛选,确保其能正确折叠成目标结构。
本研究还描述了一种重新设计环肽骨架序列的方法,与先前描述的针对相同肽骨架的Rosetta序列设计方法生成的序列相比,AfCycDesign序列显示出更高的pLDDT值和更好的折叠倾向性。通过对13残基环肽进行大规模重新设计,比较AfCycDesign序列与Rosetta设计的序列,揭示出一些关键差异,包括AfCycDesign设计增加了对疏水性氨基酸和构象受限氨基酸的使用。此外,研究人员进一步扩展了AfCycDesign方法,实现了环肽序列与结构的同步虚拟生成,并将其应用于枚举由7-13个残基组成环肽的数十万个结构聚类,最终得到了10,681个独特的聚类,这些聚类被预测能以极高的置信度(pLDDT>0.9)折叠成所设计的结构。重设计和虚拟生成环肽的X射线晶体结构解析结果,进一步证实了该方法的准确性:所有8个环肽(1个重设计环肽和7个虚拟生成环肽)的X射线晶体结构均与其设计模型极为接近,RMSD均小于1.0 Å。由于仅依赖X射线晶体学进行结构验证,因此所设计的肽可能还存在其他未结晶且未被观察到的构象。值得注意的是,通过虚拟生成方法,成功设计出11-13个残基的较大环肽;此前采用最先进方法设计这类环肽时,若不引入额外交联键则难以实现稳定,而本方法无需额外交联即可达成稳定效果。
被预测能以高置信度(pLDDT>0.9)折叠为设计结构的虚拟生成环肽及其镜像异构体,是整合靶向结合、膜穿透等功能的优良骨架。为了探究骨架与蛋白质靶点的结合能力,研究人员通过将来自p53的5 个残基短基序移植到虚拟生成环肽骨架上,设计并表征了针对MDM2的结合剂。与环肽结合的MDM2的X射线晶体结构与设计模型匹配度非常高,这证实了大环结构及其结合模式的准确性。通过计算机模拟计算表明,在先前描述的1014个热点环区中,有798个可以被移植到这组虚拟生成骨架上,并可通过设计实现与对应靶点的结合。通过设计并表征三种针对Keap1的环肽结合剂,在竞争性荧光偏振分析中显示出IC50 <200 nM,从而证实了该流程的可行性。
先前已经指出了L型和D型氨基酸排布模式对于生成具有结构性的环肽的重要性;然而,本文描述的虚拟生成的环肽突破了这一规律,尽管仅由L型氨基酸组成,它们依然能够良好折叠。本研究认可D型氨基酸和其他非标准氨基酸所提供的蛋白酶稳定性和代谢稳定性方面的优势,并且相信虚拟生成骨架中的特定位点可以进一步突变为非标准氨基酸。作为原理验证,研究人员在针对MDM2的结合剂中引入了D型氨基酸和非标准氨基酸,并证明其结合亲和力相较于原始设计有所提高。本研究未测定所报道设计产物的蛋白酶稳定性或血清稳定性,但未来针对稳定性的大规模研究数据,有望为AfCycDesign在序列设计和虚拟生成方面的优化提供指导。虽然当前版本的AfCycDesign暂不支持非标准氨基酸的设计,但正如本研究中MDM2结合剂RMG_14c的优化案例所示,可将 AfCycDesign与现有基于物理的方法结合,在设计优化中引入非标准氨基酸。此外,随着全原子深度学习模型的最新进展,本工作为未来开发深度学习网络奠定了基础,这些网络能够在骨架采样和序列设计过程中纳入更广泛的化学多样性。
当前及未来研究的重点方向之一,是改进针对治疗靶点的环肽结合剂从头虚拟生成计算方法。在过去的五年间,深度学习方法已经在治疗性蛋白质设计领域取得了巨大进展。借助本文提出的计算方法,类似的进展可以扩展到具有重要治疗价值的、结构化的环肽的定制化设计中。
https://www.nature.com/articles/s41467-025-59940-7
----------微科盟精彩文章----------
科研 | 内农大:代谢组学揭示肠道益生菌鼠李糖乳酪杆菌X9C17高产氨基酸的代谢特征(国人佳作)
科研 | 浙大&浙工大:益生菌YJ5可通过调节肠道菌群,产生有益代谢物和增强黏膜屏障来缓解便秘(国人佳作)
如果需要原文pdf,请扫描文末二维码


获取此文献原文PDF、申请加入学术群,联系您所添加的任一微科盟组学老师即可,如未添加过微科盟组学老师,请联系组学老师47,无需重复添加。
请关注下方名片