

导 语
热闹的赛道,与四个更硬的变量
如果只看数字,T细胞衔接器(T cell engager, TCE)正处在一个令人兴奋的爆发期。根据公开管线信息梳理,全球已有约12款TCE获批,从临床早期到批准阶段的管线总数约288条,其中中国约75条,血液瘤与实体瘤大致各半。靶点在扩张,格式在迭代,中国企业在快速跟进—一切都指向"赛道繁荣"这个直觉性结论。
但我们想在这篇报告里提出一个不同的判断:决定2030年前TCE竞争格局的,不是靶点名单的长度,而是四个更苛刻的变量。
第一,一款TCE或一个靶点场景,是否拥有随机对照乃至总生存(OS)获益的硬证据,而不只是单臂研究里的高缓解率。第二,它的细胞因子释放综合征(CRS)与感染负担,能否被压低到门诊可管理的水平——这直接决定它能否从末线走向前线。第三,它能否在CAR-T、抗体偶联药物(ADC)和传统抗体之外,建立一个清晰、不可替代的序贯治疗位置。第四,下一代分子格式能否真正解决抗原逃逸、T细胞耗竭和实体瘤微环境这三道结构性难题,而不只是"多一个臂、多一个靶"的工程学叙事。
围绕这四个变量,本文将把散落在证据包里的试验、标签与随访数据重新排序,用一套证据分层的方法,回答一个BD和投资人最关心的问题:在这条拥挤的赛道上,哪些结论已经被证明,哪些只是信号,哪些还只是故事。
01.
要理解TCE的机会与边界,得先回到它的物理本质。TCE的共同机制,是用一个分子的一端抓住T细胞表面的CD3,另一端抓住肿瘤相关抗原,把二者强行拉近,形成一个不依赖MHC呈递的人工免疫突触,从而触发T细胞活化、细胞因子释放和穿孔素/颗粒酶介导的杀伤。
这个机制有两个天然优势:一是不需要患者肿瘤细胞自己呈递新抗原,绕开了肿瘤免疫逃逸的一个主要环节;二是可以用现货型药物直接调动患者体内已有的T细胞,无需像CAR-T那样离体改造与回输。但同样的机制也内嵌了两个风险:过度的T细胞激活会带来CRS和免疫效应细胞相关神经毒性(ICANS);而当正常组织也低水平表达靶抗原时,会出现on-target/off-tumor毒性。
所以,TCE本质上是一门"可调强度的免疫突触工程"——如何在杀伤强度与安全窗之间找到那个可临床落地的平衡点。三个历史锚点最能说明这一点。
Blinatumomab(CD19×CD3,BiTE)是第一个用随机III期证明CD3重定向杀伤具有临床硬价值的分子:在复发难治B细胞急性淋巴细胞白血病(R/R B-ALL)中,它把中位OS从化疗组的4.0个月提高到7.7个月,完全缓解率也优于化疗[E01]。但它需要连续静脉输注,给药负担极高,代表了第一代BiTE在半衰期和便利性上的先天局限。
Catumaxomab(EpCAM×CD3,三功能抗体)走的是另一条路:通过局部腹腔给药治疗恶性腹水,把中位穿刺间隔生存(puncture-free survival)从11天延长到46天[E02]。它的价值在于提示——局部给药和局部疾病场景,可以部分规避实体瘤全身暴露的毒性难题。
Tebentafusp(gp100肽-HLA-A*02:01×CD3,ImmTAC)则证明了TCE样平台可以在实体瘤成立:在HLA-A*02:01阳性的转移性葡萄膜黑色素瘤中,它取得了1年OS 73% vs 59%、死亡风险比(HR)0.51的随机III期结果[E03]。但它的成立前提是严格的HLA限制和生物标志物入组。
这三个锚点共同说明:TCE的临床价值来自"靶点可及性、T细胞可调动性、给药场景与风险控制"的组合,而不是双抗格式本身。不是任何"肿瘤抗原+CD3"都能成药。
02.
为什么必须做证据分层:把信号和结论分开
TCE领域最大的认知陷阱,是把不同强度的证据混为一谈——用一款药的单臂ORR,去和另一款药的随机OS并列比较;或者把公司新闻稿里的早期缓解率,当作已被验证的疗效结论。要做出可靠判断,必须先给证据定级。
我们把本证据包按四级组织。A级是随机对照并显示OS、PFS或明确临床获益的证据,它是判断赛道价值的"地基",代表包括blinatumomab的TOWER、tebentafusp的III期、catumaxomab的II/III期,以及后文将重点讨论的STARGLO。B级是单臂注册或关键II期研究,足以支撑加速批准或条件批准,能证明高缓解率和可管理安全性,但难以单独证明相对标准治疗的优势——MM和B-NHL的大多数明星资产都在这一层。C级是FDA/EMA标签、审评总结和长期随访海报,用于约束真实用法、黑框风险和确认性试验义务。D级则是登记信息、公司披露和早期会议数据,只能指示开发方向和竞争态势,不能替代疗效确认。
这套分层之所以关键,是因为它直接改变结论的可信度权重。同样是"ORR超过80%",出现在3年随访的注册研究里,和出现在中位随访仅3个多月的公司资料里,含义完全不同。本文后续对每一个关键数字,都会标注其证据层级。此外需要预先声明一条贯穿全文的比较原则:除随机对照结果外,本文涉及不同研究之间的疗效/安全并列描述,均为跨试验的描述性比较,受入组人群、治疗线别、随访成熟度和终点定义影响,不能视为头对头优劣结论。
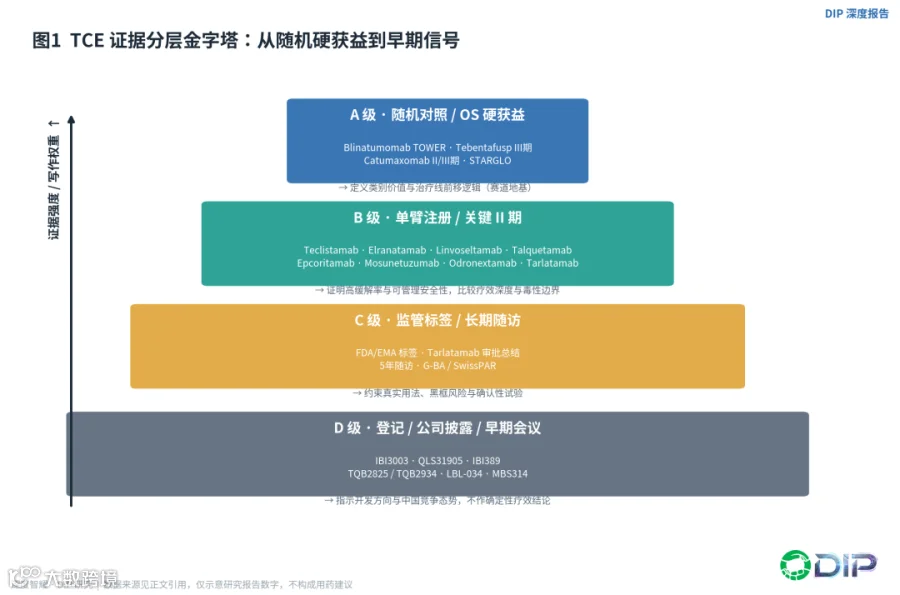
〔图1:TCE证据分层金字塔——从随机硬获益到早期信号〕
03.
为什么血液瘤先成功:可及性、谱系靶点与治疗线
TCE在血液肿瘤率先兑现,不是偶然。B细胞恶性肿瘤和多发性骨髓瘤具备几个结构性优势:靶抗原相对谱系限制(如CD19、CD20、BCMA、GPRC5D),肿瘤细胞与效应T细胞在骨髓、血液和淋巴组织中空间距离短、更容易形成突触,肿瘤负荷和微小残留病灶(MRD)可以动态监测,且末线患者存在明确且紧迫的未满足需求。
实体瘤则几乎在每一个维度上都更难:靶抗原常在正常组织有表达,药物要穿透异常血管、致密基质和高间质压才能到达肿瘤核心,而进入肿瘤的T细胞还会被PD-1/TIM-3/LAG-3等检查点、缺氧、髓系抑制细胞和TGF-β层层压制。
这个差异决定了两类肿瘤的竞争已经处在完全不同的阶段。血液瘤TCE已经进入"同靶点内差异化"的比拼——比较缓解深度、给药节奏、CRS、感染和序贯位置;而实体瘤TCE还停留在更早的"哪类靶点、哪类组织场景能被TCE转化"的验证阶段。用一句话概括:血液瘤已经完成了"药物类别确认",实体瘤还在做"靶点和场景选择确认"。
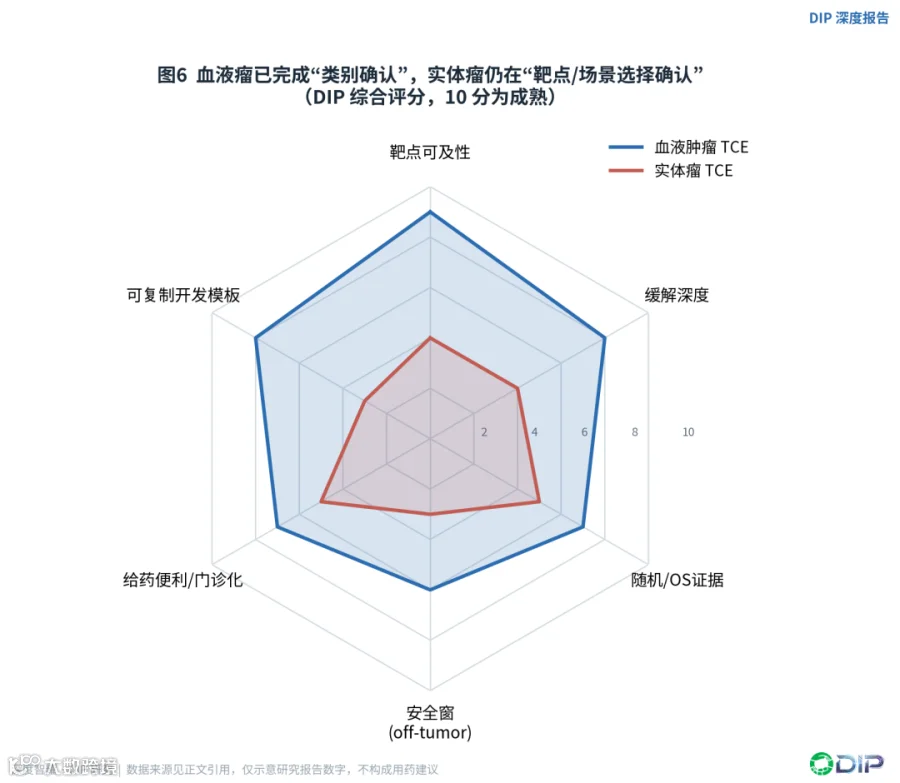
〔图6:血液瘤 vs 实体瘤TCE成熟度雷达〕
04.
多发性骨髓瘤:BCMA已拥挤,GPRC5D与双靶定义下一轮
BCMA×CD3三强:真正的差异不在ORR
Teclistamab、elranatamab和linvoseltamab三款BCMA×CD3,已经共同证明了这一靶点在经过三类药物暴露的RRMM中的高活性,但它们的差异恰恰不在缓解率本身。
Teclistamab在MajesTEC-1中ORR 63.0%、≥CR 39.4%、中位PFS 11.3个月,CRS虽高达72.1%但3级CRS仅0.6%,采用皮下每周给药(SC QW)[E04]。Elranatamab在MagnetisMM-3中ORR 61.0%、≥CR 35.0%、15个月PFS率50.9%、OS率56.7%,CRS 57.7%且无3级以上CRS,可从每周切换至每两周给药——但它的感染任意级达69.9%、3-4级达39.8%,提示BCMA TCE的长期难点远不止CRS[E05]。Linvoseltamab在LINKER-MM1的200mg组ORR 71%、≥CR 50%、中位DOR长达29.4个月,CRS约46%、3级仅0.9%,并引入"响应后每4周一次"的响应适配给药逻辑[E06]。
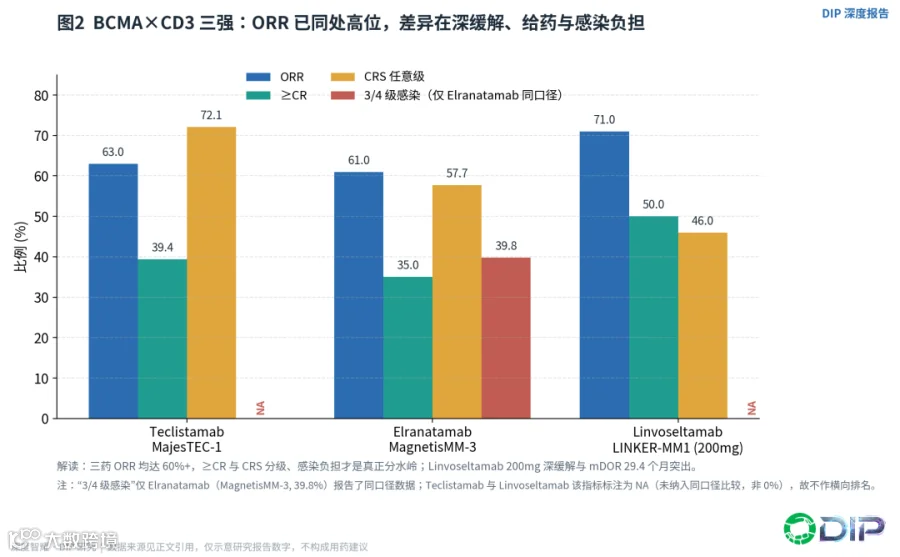
〔图2:BCMA×CD3三强疗效—安全对比(ORR/≥CR/CRS/感染)〕
把这三组数据放在一起,结论很清晰:三药ORR都已站上60%以上的高位,ORR已经不再是核心壁垒。需要提醒的是,以上比较为跨试验描述性比较,受入组人群、治疗线别、随访成熟度和统计口径影响,不能视为头对头优劣结论。真正区分它们的,是深缓解的持久性(linvoseltamab的DOR最突出)、给药节奏能否低频化门诊化,以及最关键的——感染和长期免疫抑制的管理。BCMA CAR-T已经有极高的缓解率,而ADC、CAR-T、TCE共享BCMA靶点可能造成抗原逃逸和序贯疗效衰减。TCE的核心优势是现货、可重复给药、无需细胞制备;劣势则是持续T细胞刺激带来的浆细胞相关免疫抑制和慢性感染风险。因此BCMA TCE未来的胜负手,是谁能证明更好的前线联合、固定疗程、响应适配减量、感染预防和真实世界可及性。
而在拥挤的BCMA靶点上,真正决定资产价值的其实是序贯逻辑,而非孤立的缓解率。BCMA CAR-T、BCMA ADC(如belantamab类)与BCMA TCE同靶竞争,意味着一位患者身上"谁先用、谁后用"会直接改变后续药物的疗效天花板:一旦前一款BCMA药物筛选出抗原低表达或阴性的克隆,后一款同靶药物的缓解深度和持久性都会打折。这就带来三个对BD和临床都很现实的判断维度——其一,TCE作为"现货、可重复给药"的属性,天然更适合放在CAR-T制备等待期的桥接,或CAR-T后复发的补救位置,而不是与CAR-T正面争夺一次性深度缓解;其二,若把TCE前移到CAR-T之前,就必须回答"用掉BCMA靶点是否会削弱后续CAR-T疗效"这一序贯代价;其三,正因如此,GPRC5D、双靶或BCMA后再挑战的临床数据,价值往往高于又一条ORR相近的BCMA单靶资产。换言之,BCMA赛道的竞争已经从"缓解率排名"转入"靶点排布与序贯站位"的博弈。
中国桥接数据:疗效不弱,感染管理是核心命题
中国队列进一步强化了这个判断。Teclistamab中国队列(N=26)的ORR达76.9%、≥CR 57.7%、MRD阴性14/15,12个月的DOR/PFS/OS率分别为78.5%、68.0%、83.5%——疗效不弱于甚至略高于全球数据。但感染发生率高达96.2%,其中3级以上73.1%[E08]。27.2个月的长期随访显示,ORR和深缓解得以保持,且新发3级以上感染随时间下降[E10],这为低频给药和免疫球蛋白管理的价值提供了直接支撑。
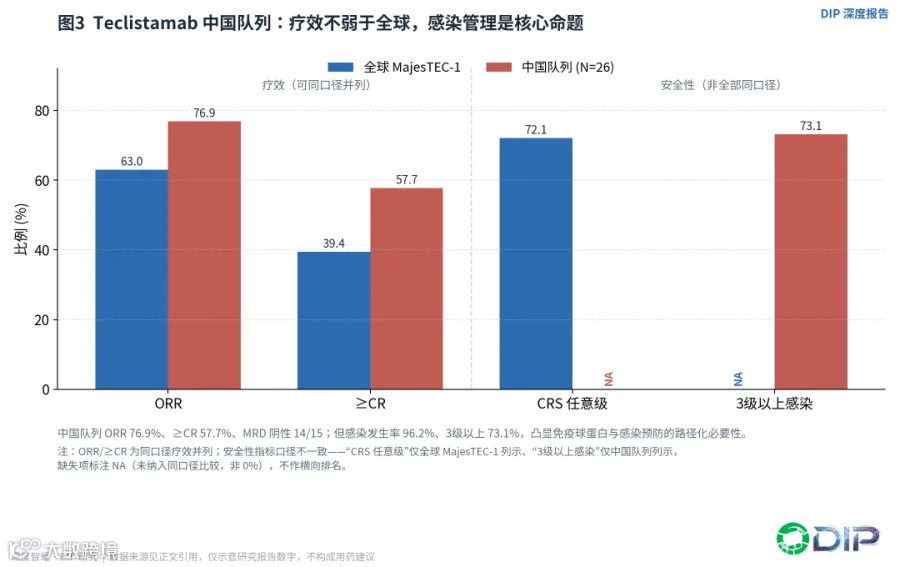
〔图3:Teclistamab中国队列 vs 全球——疗效不弱,感染管理是核心〕
中国人群这组"高疗效、高感染"的数据,恰恰是全篇核心论点的缩影:在血液瘤里,TCE的缓解能力已经不是问题,能不能把安全和可及做成路径化管理才是问题。
GPRC5D:不是替代BCMA,而是BCMA后的换靶接力
Talquetamab把GPRC5D确认为MM中第二个成熟TCE靶点。全球MonumenTAL-1数据显示,推荐剂量下ORR约64%-70%,CRS常见但多为1/2级[E07]。它更关键的价值在于GPRC5D表达不依赖BCMA,能在BCMA失败或BCMA压力下提供换靶路径。中国队列中,0.4mg/kg每周组ORR 69.0%、≥CR 37.9%、中位DOR 15.7个月、中位PFS 8.3个月;0.8mg/kg每两周组ORR 66.7%、≥CR 50.0%,DOR/PFS未达[E09]。
GPRC5D的差异化毒性值得单独强调。BCMA TCE的长期风险主要指向感染和免疫球蛋白下降;GPRC5D则叠加了口腔、味觉、皮肤、指甲和体重下降等on-target/off-tumor问题。这类毒性不一定导致早期停药,却会实实在在地影响长期依从性、营养状态和门诊管理成本。所以GPRC5D的准确定位不是"BCMA耐药后的最佳接力"这么简单,而是以一套不同的毒性谱,换取靶点独立性。
双靶/三特异性:理论优势强,但仍是D级证据
BCMA×GPRC5D双靶或三特异性,希望同时降低单靶抗原逃逸、提高BCMA经治和髓外病变人群的响应。IBI3003的公司资料显示,在剂量≥120μg/kg的24例可评估患者中ORR达83.3%,髓外病变ORR 80%,既往BCMA和/或GPRC5D治疗患者ORR 77.8%,CRS均为1/2级,并获得FDA快速通道资格[E24]。
这是非常亮眼的早期信号,但必须清醒地标注:这属于D级证据——样本量小、中位随访仅3.25个月、数据来自公司披露,不能替代疗效确认。工程学故事只有转化为经随访验证的持久缓解,双靶/三特异性才会从"更好的分子设计"真正进入"临床差异化"。
对国产BCMA×CD3的me-too策略而言,窗口正在收窄。据公开信息整理,NMPA引入进口BCMA×CD3产品的时间窗约19-20个月,这意味着国产资产若只有相似ORR,很容易被进口先发、医保谈判和CAR-T竞争同时挤压。更可行的突围路径是:瞄准后BCMA人群、切GPRC5D或双靶、做低频/固定疗程/门诊化,或与CD38单抗/免疫调节剂构成前线组合。
05.
CD20×CD3:真正的分水岭是STARGLO,而不是单药获批
CD20×CD3的竞争比BCMA更复杂,因为B细胞非霍奇金淋巴瘤(B-NHL)涵盖弥漫大B细胞淋巴瘤(DLBCL)、滤泡性淋巴瘤(FL)、转化FL、CAR-T后复发、老年/不适合移植等多个截然不同的场景。这里的差异不是简单的ORR比较,而是给药方式、疗程长度、联合策略、FL与DLBCL定位,以及CAR-T前后序贯。
Glofitamab与STARGLO:从末线单药到随机联合
Glofitamab单药在NP30179中采用obinutuzumab预处理和12周期固定疗程治疗R/R DLBCL,CR率39%、12个月PFS率37%、CRS 63%且3级以上仅4%[E11]。这证明了固定疗程的CD20×CD3可以在高度难治的DLBCL中产生深缓解。
但真正的分水岭是STARGLO。glofitamab+GemOx对比R-GemOx,在不适合自体移植的R/R DLBCL中给出了随机III期证据:约2年随访时,24个月OS率为54.4% vs 33.6%,OS、PFS、CR率均持续优于对照组[E12][E13],3年更新进一步强化了生存获益方向[E14]。
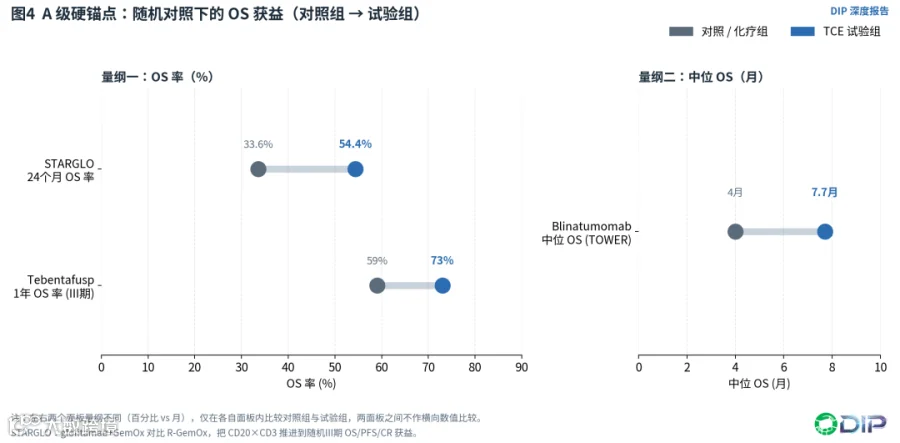
〔图4:A级硬锚点——随机对照下的OS获益(STARGLO/TOWER/Tebentafusp)〕
STARGLO的深层意义有三点。第一,它把TCE从"末线单药缓解"推进到"化疗骨架上的免疫增强联合",是CD20×CD3迄今最强的证据锚点,也是整个TCE血液瘤领域少数几个A级证据之一。第二,它提示TCE在前移治疗线时,可能更适合作为组合模块,而不是单药替代全部标准疗法。第三,它把ctDNA动态、治疗后免疫恢复、固定疗程后缓解维持纳入讨论,使CD20×CD3的竞争从"ORR竞赛"升级为"疗程结束后能否维持免疫和缓解"的问题。
Epcoritamab、Mosunetuzumab、Odronextamab:场景分化
Epcoritamab在EPCORE NHL-1中治疗R/R LBCL,ORR 63.1%、CR 38.9%、中位DOR 12.0个月,CRS 49.7%且多为1/2级,采用皮下给药并逐步从每周转向每两周、每四周[E15]。它的差异化是给药便利性和联合开发空间,但如果治疗持续到进展或不可耐受,长期感染、门诊监测和总治疗成本就会成为真实世界的关键变量。
Mosunetuzumab在R/R FL中展现了另一种逻辑——不追求DLBCL里的强联合,而是在惰性淋巴瘤中建立有限疗程、深缓解和免疫恢复。GO29781长期数据中,ORR约78%-80%、CR约60%,5年随访显示整体人群5年OS约78.5%,CR患者5年OS约91.6%,中位至下次治疗时间(TTNT)约64.1个月,且固定疗程后B细胞和免疫球蛋白恢复[E16]。对FL而言,这类数据比短期ORR重要得多——患者预期生存长,治疗目标是延长无治疗间隔、降低长期免疫抑制、保留后续治疗选择。
Odronextamab在CD20阳性B-NHL中活性明确,ELM-2的FL 3年随访显示ORR 80.5%、CR 74.2%、中位完全缓解持续时间(DOCR)35.2个月,CR患者中位PFS 38个月、OS未达[E17]。但它在美国经历的完全回复函(CRL)提示:TCE的成败不只由缓解率决定,还受生产、剂量优化、感染预防、给药复杂度和监管可接受性影响。高活性不等于顺利商业化,监管科学会实质影响最终竞争力。
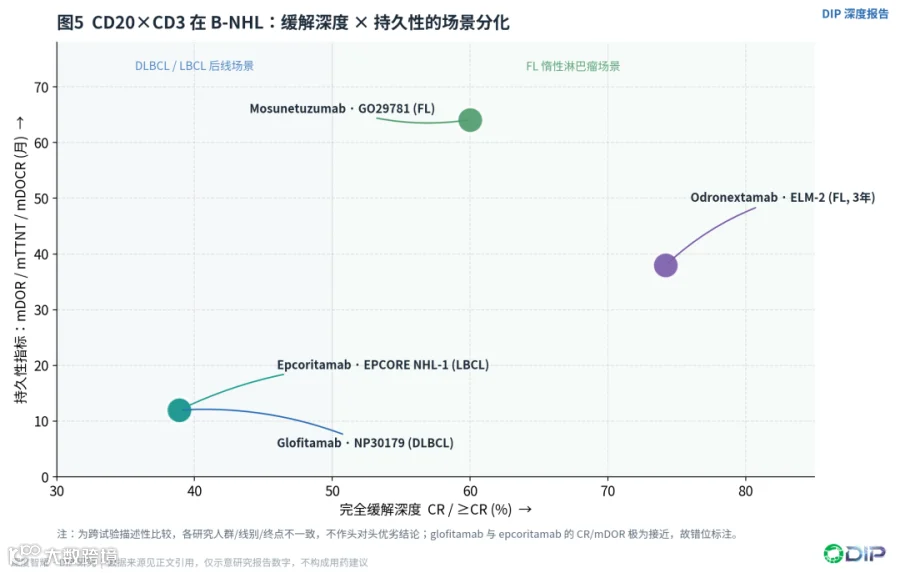
〔图5:CD20×CD3在B-NHL的缓解深度×持久性定位矩阵〕
综合来看,CD20×CD3已经出现清晰的场景分化:DLBCL场景(glofitamab、epcoritamab)比拼的是难治后线的深度缓解、随机联合证据和给药便利;FL场景(mosunetuzumab、odronextamab)比拼的是固定疗程后的深缓解、超长无治疗间隔和免疫恢复。同样需要强调,上述对多款资产的并列描述属于跨试验比较,各研究在疾病亚型、治疗线别、随访时长和终点定义上并不一致,不能据此直接排名优劣。
更进一步看,DLBCL与FL两个场景的差异不仅在于数据,更在于商业化路径和临床采用逻辑。DLBCL是侵袭性疾病,患者需要快速、深度的缓解,因此固定疗程带来的"停药后维持缓解"叙事、以及STARGLO式的随机联合证据,会直接转化为进入指南和一线/二线骨架的谈判筹码;同时DLBCL患者往往与CAR-T争夺同一批人群,CD20×CD3必须清晰回答"CAR-T前桥接、CAR-T后复发、还是不适合CAR-T的替代"这三种定位中自己站在哪里。FL则是惰性、可反复复发的长病程疾病,患者预期生存以年计,治疗目标不是一次性治愈,而是尽可能延长无治疗间隔、把长期免疫抑制和感染负担压到最低——这决定了固定疗程、缓解后免疫恢复、门诊可执行性,比短期ORR更能决定FL资产的真实价值。对商业化而言,DLBCL拼的是"证据强度与骨架卡位",FL拼的是"长期可管理性与治疗节奏",两者需要完全不同的临床开发与市场进入策略。
06.
DLL3与实体瘤:Tarlatamab是突破口,但不能外推
Tarlatamab是实体瘤TCE最重要的临床突破。在DeLLphi-301中,10mg每两周组ORR 40%、中位PFS 4.9个月、9个月OS率68%、CRS 51%且3级仅1%;而100mg组ORR更低、3级CRS更高,支持10mg作为更优风险收益剂量[E18]。FDA审批总结进一步明确:获批剂量为1mg step-up后10mg每两周,99例患者ORR 40%、中位DOR 9.7个月,标签带有严重/危及生命CRS和神经毒性的黑框警告,加速批准需要DeLLphi-304等随机研究确认[E19]。
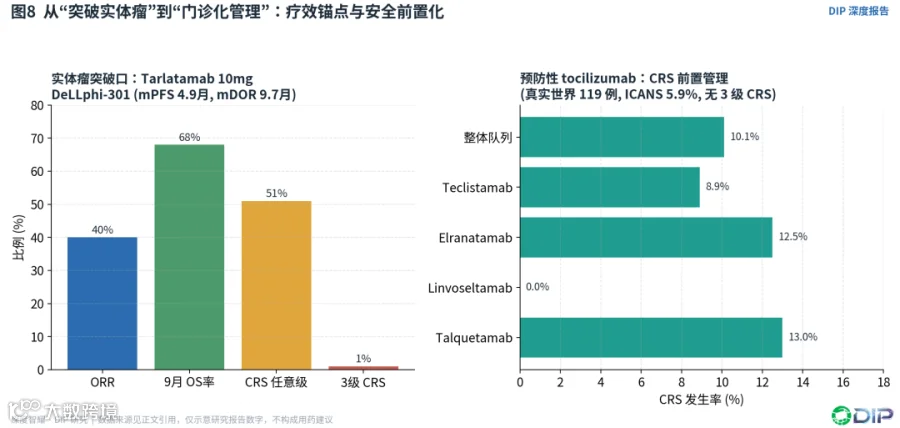
〔图8:实体瘤突破口Tarlatamab与预防性tocilizumab门诊化〕
Tarlatamab成功的关键,不是"实体瘤也能做TCE"这句口号,而是DLL3具备相对特殊的成药条件:它在小细胞肺癌(SCLC)的神经内分泌肿瘤细胞表面高表达且异常定位于细胞表面,而在正常组织中多为低表达或位于细胞内不可及。这种"肿瘤细胞表面可及、正常组织不可及"的组合,才是实体瘤TCE的核心门槛。
正因如此,tarlatamab的成功不能简单外推。实体瘤TCE目前面临三重障碍:其一是抗原异质性,实体瘤容易出现抗原阴性亚克隆,单靶TCE可能诱导选择性逃逸;其二是物理与代谢屏障,异常血管、致密基质、高间质压、缺氧会阻挡药物和T细胞进入肿瘤核心;其三是T细胞功能障碍,持续CD3刺激带来耗竭,TME中的髓系抑制、Treg、TGF-β、腺苷和检查点通路会削弱杀伤[E21]。CLDN18.2、PSMA、MUC17、GPC3、B7-H3等每一个靶点,都需要各自回答表达阈值、正常组织安全窗、抗体可及性和TME抑制的问题。
中国实体瘤TCE中,QLS31905值得单列。据登记信息,它正在开展QLS31905联合白蛋白紫杉醇/吉西他滨对比安慰剂加化疗的一线CLDN18.2阳性晚期胰腺癌III期研究[E25],这是中国TCE从血液瘤向实体瘤随机验证迈进的关键样本。IBI389则是CLDN18.2/CD3方向的Ia/Ib研究,探索单药、联合信迪利单抗和/或化疗[E26]。需要强调的是,这两者均属登记/早期层面证据,不能替代疗效确认。它们真正的看点不是"进入III期"本身,而是能否证明在化疗/免疫治疗基础上叠加TCE后带来OS或PFS增益,且不因胃肠道正常组织表达CLDN18.2而产生不可接受的毒性。
07.
安全性:CRS已可控,感染与长期免疫抑制才是前线的硬门槛
早期TCE最大的恐惧是CRS。而现在的现实是:阶梯剂量、预处理、分次给药、住院观察和tocilizumab/激素管理,已经把3级以上CRS控制到较低水平。真正未被解决的,是低级别CRS仍会占用住院观察和急诊资源、拉低患者体验——而当TCE向前线推进时,患者对安全性和便利性的容忍度会显著下降。
预防性tocilizumab是一个有前景的方向。一项真实世界研究中,119例MM患者在首个step-up剂量前接受单剂tocilizumab预防,整体CRS率降至10.1%、ICANS率5.9%、无3级CRS;teclistamab、elranatamab、linvoseltamab、talquetamab对应的CRS率分别为8.9%、12.5%、0%和13%[E20]。这提示CRS管理可能从"反应式救治"转向"前置预防",并为门诊step-up的探索打开空间。
但感染问题更顽固。BCMA和CD20相关TCE会造成浆细胞或正常B细胞耗竭、低免疫球蛋白血症和机会性感染风险。elranatamab、teclistamab中国队列、mosunetuzumab和odronextamab的长期数据,都反复指向同一个结论:感染、IgG下降、静脉注射免疫球蛋白(IVIG)使用、PJP/病毒预防和疫苗策略,必须成为TCE治疗方案的内在组成部分,而不是标签里的附属条款[E05][E08][E16][E17]。对前线联合尤其如此——若TCE与CD38单抗、免疫调节剂、化疗或PD-1/PD-L1联用,免疫抑制和感染的叠加风险可能抵消疗效收益。
为什么说感染而非CRS才真正决定TCE能否前移?因为CRS是急性、可预期、可在数天内用tocilizumab和激素处置的事件,本质上是"一次性、可预案"的风险;而感染是慢性、累积、贯穿整个持续给药周期的风险,随暴露时间延长而叠加,且在早线、体能状态更好、预期用药更久的人群里,长期免疫抑制的代价会被进一步放大。末线患者可以为高缓解率承受高感染负担,但一位有数年预期生存的早线患者,很难接受长期反复的机会性感染和住院——这正是感染管理能否路径化,决定TCE前移天花板的根本原因。
把这一判断落到临床操作层面,可以拆成一个相互绑定的管理框架。其一,门诊step-up的前提条件:需要预防性tocilizumab把早期CRS压到低位、明确的分级监测与急诊回流通道、以及经过培训的社区/门诊团队,三者缺一不可,否则"现货"的便利优势会被住院观察成本吞掉。其二,IVIG介入的必要性:BCMA/CD20类TCE持续耗竭浆细胞或B细胞,低免疫球蛋白血症几乎是必然结果,IVIG不应是感染发生后的补救,而应作为按IgG阈值触发的常规支持,纳入方案设计。其三,疫苗与感染预防前置:PJP预防、疱疹/巨细胞病毒等病毒预防、以及在免疫抑制加深前尽早完成的疫苗接种,都必须"前置"到治疗启动阶段——一旦B细胞耗竭,疫苗应答会显著下降。其四,固定疗程/减频给药与安全管理绑定:固定疗程和响应适配减量之所以重要,不只是便利,而是通过缩短暴露时间直接降低累积感染和免疫抑制负担,让"停药后免疫恢复"成为可能。这四条不是各自独立的注意事项,而是一套必须整体设计的落地体系。
长期看,TCE的安全竞争会从"有没有3级CRS"转向四个更实际的问题:能不能在门诊完成step-up;能不能固定疗程停药;停药后免疫是否恢复;感染、神经毒性、皮肤/味觉/体重下降等慢性毒性是否影响生活质量。谁能把这些问题做成路径化管理,谁才更可能从末线市场走进前线。
08.
下一代TCE:六条工程化路线,各有各的代价
下一代TCE不是简单地"多一个靶点、多一个共刺激臂",而是在疗效、安全和可制造性之间重新分配风险。可以归纳为六条路线,每一条都有明确的解决目标,也都有各自的代价。
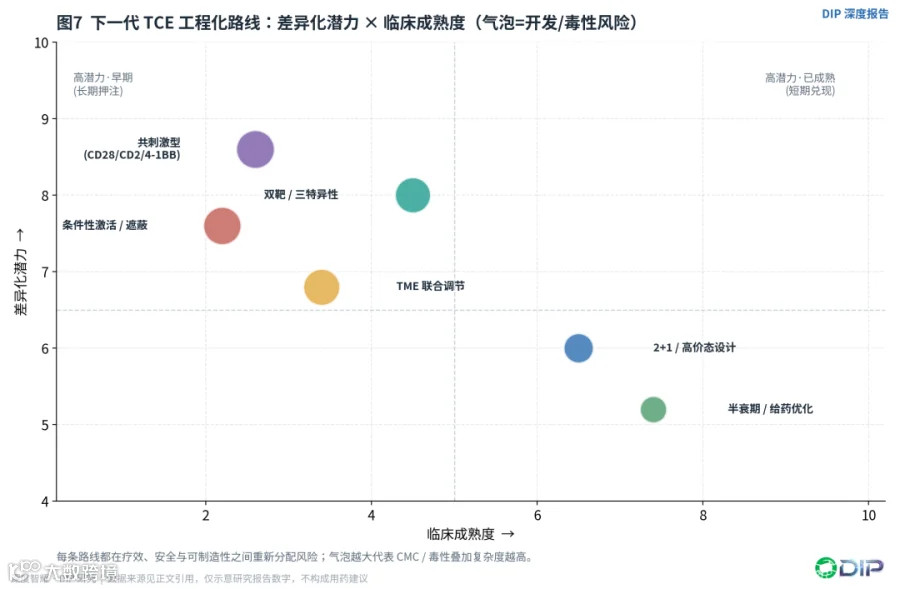
〔图7:下一代TCE工程化路线——差异化潜力×临床成熟度〕
第一,2+1或高价态设计。通过增加肿瘤抗原结合臂提高对高抗原密度肿瘤细胞的选择性,同时降低对低表达正常组织的结合。glofitamab的2:1 CD20:CD3结构和若干国产高价态管线属于此类。代价是分子更复杂、PK和制造更难,且高亲和/高价态本身可能提高局部或全身激活。这条路线临床成熟度相对最高。
第二,双靶/三特异性。BCMA×GPRC5D×CD3等试图降低抗原逃逸并覆盖BCMA后人群。但多靶并不自动等于更高疗效——若两个肿瘤抗原表达不共定位,或正常组织表达叠加,反而可能扩大毒性窗口。潜力大,但需随访成熟度兑现。
第三,共刺激型TCE。CD28、CD2、4-1BB等共刺激信号,用于弥补单一CD3信号导致的T细胞无能/耗竭。非超激动CD28双信号研究显示,肿瘤抗原定位的CD28共刺激可与CD3 TCE协同增强T细胞激活,同时避免CD28的系统性超激动[E22];CD2共刺激平台(如EVOLVE)则将signal 2整合进TCE,以提高实体瘤和血液瘤的杀伤[E23]。核心风险是细胞因子和系统性免疫激活,必须证明共刺激被严格限制在肿瘤抗原依赖的场景。这条路线差异化潜力最高,但临床最不成熟。
第四,条件性激活/遮蔽型TCE。通过TME蛋白酶、酸性pH或局部条件解遮蔽CD3或肿瘤抗原结合臂,扩大实体瘤的正常组织安全窗,PSMA、EGFR、B7-H3等靶点尤其需要。代价是疗效依赖TME酶活和空间分布,开发复杂度陡增。
第五,半衰期和给药优化。从连续输注走向IgG样TCE和每周/每两周/每四周给药,甚至响应适配给药。给药便利性不是商业细节,而是能否进入社区、门诊和前线治疗的前提。
第六,TME联合调节。实体瘤TCE很可能需要与PD-1/PD-L1、TGF-β抑制、VEGF/血管正常化、髓系重塑、放化疗联用。单纯提高CD3激活强度无法解决肿瘤内T细胞排斥,反而可能增加外周毒性;理想方案应先用生物标志物筛选"抗原阳性、T细胞可进入、免疫抑制可逆"的人群。
09.
中国TCE竞争格局:管线数量不是壁垒
据公开信息整理,中国临床至批准阶段的TCE管线约75条,血液瘤和实体瘤大致各半。这说明中国企业已成为TCE领域的全球重要参与者、在部分方向具备前排竞争力,但也意味着拥挤和同质化风险极高——BCMA×CD3、CD20×CD3和GPRC5D×CD3是最容易陷入价格战和适应症重叠的方向。
我们把中国管线分为四类。进度型fast-follow:国产BCMA/CD20/GPRC5D×CD3进入报产或III期,优势是上市速度和本土执行,风险是进口先发已完成市场教育且疗效差异有限。这里的时间窗值得算细账:据公开信息整理,NMPA引入进口BCMA×CD3产品的时间窗约19-20个月,这段"先发差"看似不长,却足以让进口产品率先完成大型中心的准入、医生用药习惯的培养和首轮医保谈判——等国产me-too上市时,定价权、中心渠道和医生教育红利大多已被锁定,只能依靠价格竞争,利润空间被大幅压缩。因此单纯"更快跟进"很难构成壁垒。机制型fast-better:IBI3003、MBS314、TQB2934等双靶或三特异性,希望在BCMA后、髓外病变或高危人群做出差异,成败取决于成熟随访而非早期ORR。需要清醒的是,双靶/三特异性若不能在后BCMA、髓外病变、高危细胞遗传学等真正未满足的人群中给出经随访验证的持续优势,就难以摆脱"只是更复杂、更贵的me-too"这一质疑,工程学复杂度本身不等于临床差异化。实体瘤先发验证:QLS31905的CLDN18.2胰腺癌III期[E25]、IBI389[E26]等,是中国实体瘤TCE能否跨过随机验证门槛的关键样本(均为登记/早期证据)。平台工程型:CD28/CD2/4-1BB共刺激、条件性激活、mRNA/局部递送、TCR样TCE,更适合做全球差异化,但需要更强的转化医学和安全模型。

〔图9:中国TCE竞争格局四象限〕
到2030年,国产TCE的存活逻辑大概率不是"谁管线最多",而是谁能在以下任一维度建立不可替代性:国产首发并快速纳入医保;在进口失败或经治人群中证明疗效;以皮下/低频/门诊路径显著降低交付成本;在实体瘤随机III期中证明OS/PFS;或通过双靶/共刺激/条件激活拿到国际监管认可。管线数量本身不构成壁垒,证据质量和适应症选择才是。
10.
2030前的五个关键临床问题
其一,MM的一线和早线能否接受持续TCE暴露。teclistamab+daratumumab等前线联合若阳性将极大扩展市场,但早线患者对长期感染、二次肿瘤、疫苗反应和生活质量的容忍度更低。TCE不能只用末线ORR去证明早线价值。什么数据会支持或推翻当前判断?若前线联合在给出PFS/OS获益的同时,能把3-4级感染率、感染相关住院和治疗中止率控制在与现有标准方案相当的水平,且固定疗程后可观察到免疫恢复,就支持TCE前移;反之,若疗效获益以显著抬升的长期感染负担和生活质量下降为代价,早线价值就会被证伪。
其二,CD20×CD3是否会重塑DLBCL一线。STARGLO已证明二线及以上的联合价值,下一步一线联合研究将决定TCE能否进入标准免疫化疗骨架。若一线获胜,CD20×CD3就会从"CAR-T后或不适合CAR-T的替代方案"变成核心治疗模块。什么数据会支持或推翻当前判断?关键看一线随机研究能否在R-CHOP类骨架之上给出无争议的PFS乃至OS增益,且安全性可门诊管理;若一线联合仅改善缓解率而未转化为生存获益,或毒性叠加抵消收益,CD20×CD3就会被限定在复发难治场景。
其三,SCLC中DLL3路径能否完成确认性试验。tarlatamab已经打开实体瘤TCE的大门,但加速批准需要随机研究确认。若DeLLphi-304等显示OS获益,DLL3将成为实体瘤TCE最强锚点;若失败,实体瘤TCE会回到"单臂缓解率不足以改变范式"的审慎状态。什么数据会支持或推翻当前判断?确认性随机研究相对化疗的OS获益是分水岭——阳性则DLL3成为实体瘤TCE的范式锚点,阴性或仅有PFS而无OS改善,则会重新质疑单臂缓解率能否预测实体瘤生存获益。
其四,实体瘤TCE能否形成可复制的开发模板。CLDN18.2、PSMA、GPC3、MUC17、EGFR都不能照搬DLL3,每个靶点都需要单独定义抗原阳性阈值、表达均一性、可及性、正常组织风险和联合策略。什么数据会支持或推翻当前判断?若在DLL3之外,至少再有一个实体瘤靶点(如CLDN18.2)跑出随机OS/PFS获益并明确安全窗,就说明存在可迁移的开发逻辑;若多个靶点反复止步于早期缓解信号而无法转化为随机获益,则说明tarlatamab更可能是DLL3生物学的特例而非模板。
其五,真实世界运营能否从大型中心下沉。TCE若长期依赖住院step-up、复杂监测和频繁输注,其"现货"优势会被运营成本抵消。预防性tocilizumab、固定疗程、每两周/每四周响应适配、IVIG路径、感染预防和远程监测,将共同决定TCE的商业化上限。什么数据会支持或推翻当前判断?若门诊step-up的真实世界安全数据(CRS/ICANS急诊回流率、感染住院率)证明可在社区中心安全执行,则下沉成立;若严重不良事件仍高度依赖大型中心的即时处置能力,TCE的可及性和商业化天花板就会被运营成本封死。
结语:终局不是"更多靶点",而是"更少但更硬的可验证场景"
TCE已经在血液肿瘤建立了牢固的类别地位:CD19、BCMA、GPRC5D、CD20都有明确的临床价值,部分资产开始向前线和联合推进。实体瘤方面,tebentafusp和tarlatamab证明TCE样平台并非无效,但可复制性仍取决于靶点生物学和工程化安全窗。中国管线丰富,部分资产在双靶/三特异性和CLDN18.2实体瘤方向具备全球竞争潜力,但管线数量本身不会自动转化为壁垒。
我们的核心判断是:2030年前,TCE的竞争会从"谁先获批"转向"谁能用高等级证据证明特定场景的净获益"。在血液瘤,净获益由深缓解、固定疗程、感染控制和序贯位置共同决定;在实体瘤,净获益由抗原可及性、TME可逆性、条件性激活和随机OS/PFS决定。
换言之,真正能活到终局的TCE,不一定是靶点最多的那个,而是能把疗效、安全、给药、监管和支付整合成一条可复制治疗路径的那个——前提是随机证据、可管理的安全性和清晰的序贯位置能够被真实兑现。赛道的终局不是"更多靶点",而是"更少但更硬的可验证场景"。
证据分级与关键引用说明
本报告严格区分证据强度,避免将不同层级的数据并列解读。分级标准与关键证据对应如下:
A级(随机对照/OS获益,赛道地基):[E01] blinatumomab TOWER随机III期(R/R B-ALL,OS 4.0→7.7个月);[E02] catumaxomab恶性腹水随机II/III期(穿刺间隔生存11→46天);[E03] tebentafusp III期(葡萄膜黑色素瘤,1年OS 73% vs 59%,HR 0.51);[E12][E13][E14] STARGLO随机III期及2/3年随访(glofitamab+GemOx,24个月OS 54.4% vs 33.6%)。
B级(单臂注册/关键II期,支撑加速或条件批准):[E04] teclistamab MajesTEC-1;[E05] elranatamab MagnetisMM-3;[E06] linvoseltamab LINKER-MM1;[E07] talquetamab MonumenTAL-1;[E11] glofitamab NP30179;[E15] epcoritamab EPCORE NHL-1;[E16] mosunetuzumab GO29781;[E17] odronextamab ELM-2;[E18] tarlatamab DeLLphi-301。
C级(监管标签/长期随访,约束真实用法与风险):[E10] MajesTEC-1中国队列长期随访;[E19] tarlatamab FDA审批总结(含黑框与确认性试验要求);及[E16][E17]所含长期随访海报。
区域桥接/补充临床证据(B/C级补充,非独立第五级):[E08] teclistamab中国队列、[E09] talquetamab中国队列,属注册药物的区域桥接研究。需要强调的是,这一类并非在A/B/C/D之外新增的第五级,而仅作为B/C级证据的补充说明——其证据强度介于B级单臂注册与C级长期随访之间,可用于校准中国人群疗效与安全的外推,但因样本量有限,不单独作为随机获益结论。
D级(登记/公司披露/早期会议,仅指示方向,不替代疗效确认):[E24] IBI3003公司资料(中位随访仅3.25个月);[E25] QLS31905胰腺癌III期登记;[E26] IBI389 Ia/Ib登记。
机制与工程化转化证据:[E20] 预防性tocilizumab真实世界研究;[E21] 实体瘤TCE屏障与条件性激活综述;[E22] 非超激动CD28共刺激;[E23] CD2共刺激平台。
关键证据对应简表
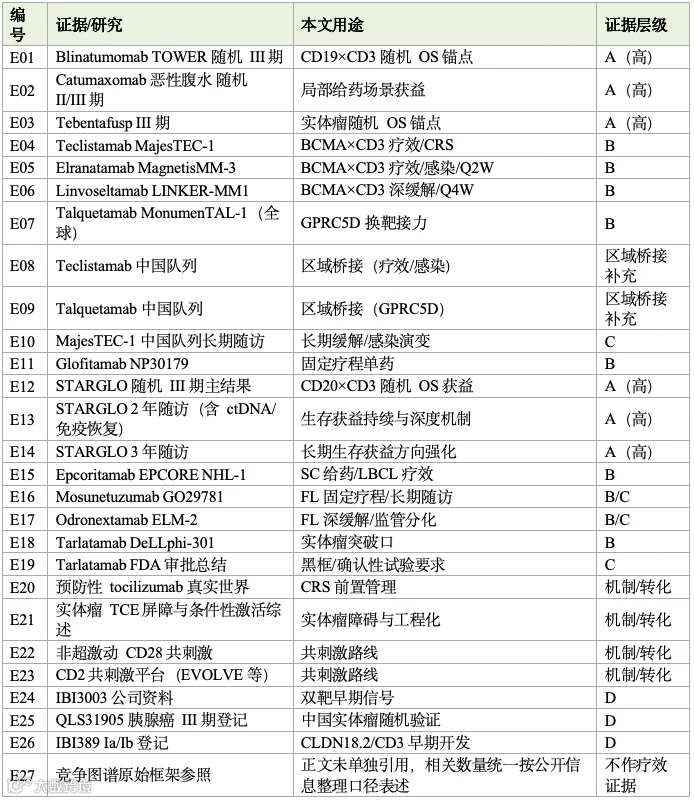
竞争图谱相关数量(全球获批约12款、全球临床至批准阶段约288条、中国约75条、NMPA引入进口BCMA×CD3产品时间窗约19-20个月)均据公开管线信息梳理整理,统一按公开信息整理口径表述,未在正文单独引用来源条目。本报告基于一份已通过质控的118篇PDF证据包(编号[R001]-[R118],含随机III期、单臂注册、FDA/EMA标签、长期随访海报及公司/登记信息),完整参考文献清单可备索。
本文所有关键数字均来自上述证据包,力求无数据幻觉;文中定性判断为研究团队独立观点,不构成任何投资或用药建议。
END

