


前言
随着中国医药企业“走出去”步伐持续加快,泰国凭借其在东南亚的战略地位、庞大的人口基数及不断扩大的医疗支出规模,正成为越来越多中国药企布局区域市场的优先目的地。许多企业已通过泰国本地合作方或注册代理,着手推进药品在泰国的注册工作;接下来面临的关键环节,是从中国完成药品出口并实现在泰国的合规销售。
泰国药品监管体系以《药品法》(Drug Act B.E. 2510,即公元1967年)为核心支柱,配合多项部颁法令和TFDA指引共同运作。由于该体系在准入路径、机构许可、逐批通关、药品分类和标签语言等方面与中国《药品管理法》体系存在显著差异,熟悉国内药品法规的业务决策者,有必要快速建立对泰国核心监管框架的系统认知,以确保合规操作、避免清关延误或法律风险。

监管体系总览
1.1 主管机构:TFDA
泰国药品监管的核心主管机构为泰国食品药品监督管理局(Thai Food and Drug Administration,简称TFDA),隶属于公共卫生部(Ministry of Public Health,MOPH)。TFDA下设药品监管司(Medicines Regulation Division / Drug Control Division),负责药品的注册、许可证签发、市场监督及药物警戒全链条管理。
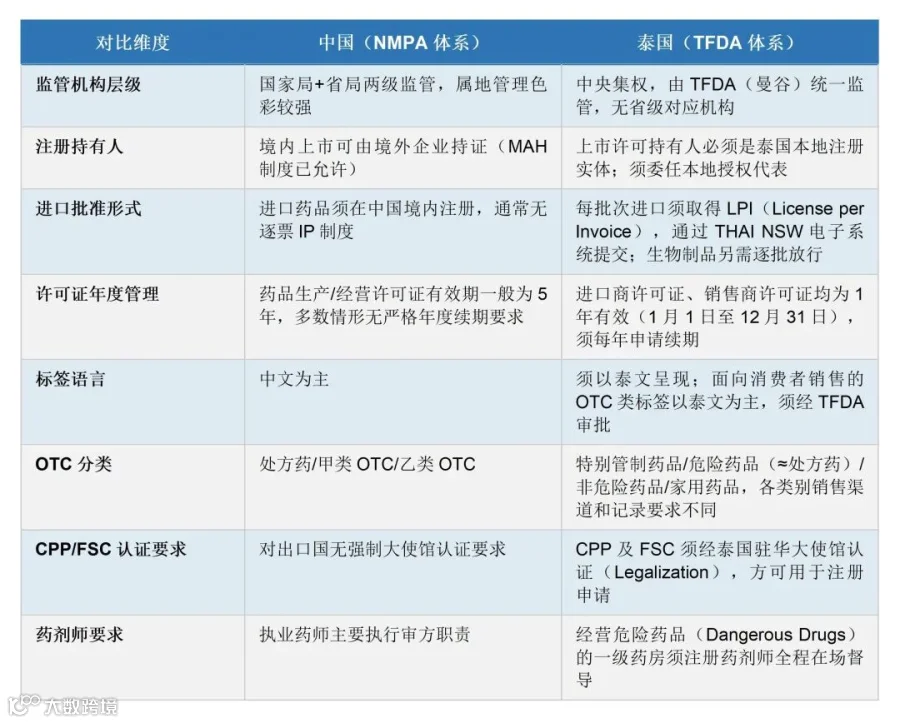
与中国对应机构类比:TFDA的职能定位与中国NMPA类似,均承担药品全生命周期的注册与监管职责;但泰国为中央集权式监管架构,不设省级对应机构,全国药品监管事项统一由TFDA(曼谷)处理。
1.2核心法规框架


药品注册制度(Drug Registration)
2.1 注册是一切合规行为的前提
药品法》第79条规定,任何人在制造或进口/订购药品入境前,须持有效药品配方注册证书(Certificate of Drug Formula Registration)。该注册证书等同于其他司法辖区通常所指的上市许可(Marketing Authorization);在《药品法》第6号修正案B.E. 2562(2019)通过后,亦正式引入"上市许可"概念并规定其7年有效期及续期制度。
2.2 注册持有人与中国制药企业的关系
在泰国监管体系下,上市许可的持有人(License Holder)须为在泰国注册的本地实体,通常为中国制药企业委托的本地注册代理或合作分销商。中国制药企业作为实际生产商(Manufacturer)在注册档案中备案,但对TFDA直接负责的是泰国本地许可持有人。

进口环节的监管要求
3.1 机构许可:进口商许可证(Import License)
依据《药品法》第12条,任何人未持有效许可证,不得在泰国境内制造、销售、进口或订购现代药品入境。从事药品进口业务的机构,须向TFDA申请并持有:
● 进口商许可证(Importer's License):持证方可进口已注册药品并在境内批发/销售;
● 销售商许可证(Drug Seller's License):根据经营药品类别不同,持证条件有所差异,经营「危险药品」(Dangerous Drugs,即处方药类)须由注册药剂师常驻并督导;
以上许可证均须每年在规定期限内(1月1日至12月31日)向TFDA申请续期,逾期将导致许可失效。
3.2 逐票通关:License per Invoice(LPI)制度
泰国对受TFDA管辖的药品实行「逐票通关」机制,每批次进口须取得TFDA核发的License per Invoice(LPI)。LPI是针对特定发票/批次货物的进口通知文件,须在海关申报前通过泰国国家单一窗口系统(THAI NSW)电子提交,并与海关进出口申报单(Import/Export Declaration)关联。

3.3 逐批放行(Lot Release)——生物制品专项要求
人用疫苗及血浆来源产品在进入泰国市场之前,须经泰国医学科学部生物制品研究所(Institute of Biological Products, Department of Medical Sciences)完成逐批质量检测,取得批放行证书(Certificate of Lot Release)后方可销售。该程序独立于LPI之外:
● 化学合成药品:一般无强制逐批放行要求;
● 生物制品(疫苗、血液制品等):批放行为强制程序,通常须提交批次生产记录、检验报告、原始批放行文件,审批周期数周至数月;
● 供应链规划须为生物制品的批放行程序预留充足时间,避免LPI+批放行双重流程叠加导致库存断供。
3.4 中国出口侧配合要求
从中国出口药品至泰国,须配合提供以下文件:
● 药品生产许可证/出口商销售许可证:证明出口方具备合法生产资质;
● 药品出口证明(Certificate for Export)或自由销售证明(Free Sale Certificate,FSC):由中国NMPA或其授权机构出具,并须经泰国驻华大使馆认证(Legalization);
● 批检验报告(Certificate of Analysis,CoA):与进口批次对应;
● 产品证书(Certificate of Pharmaceutical Product,CPP):CPP格式须符合ASEAN Harmonization要求,并须经泰国大使馆认证;
● 原产地证书(Certificate of Origin,CoO):通常由中国贸促会或商检机构出具;
● GMP证明文件:制造商GMP合规证明,PIC/S或ASEAN体系认证成员的制造商可享简化要求(2024年改革草案中)。
3.5 泰国进口清关文件要求(海关申报)
进口药品清关须通过泰国海关系统提交进口报关单(Import Entry),主要配套单证包括:商业发票、装箱单、提单/空运单、TFDA核发的LPI(通过THAI NSW电子关联)、批检验报告(CoA)、原产地证书(CoO)、自由销售证明(FSC,须经大使馆认证)、含麻醉品/精神药品成分者须附专项进口许可。
建议委托泰国持牌报关行(Licensed Customs Broker)操作清关事宜,以确保HS编码归类准确、THAI NSW系统操作合规,避免因申报失误导致货物被扣押。

流通与销售环节的监管要求
4.1 药品四类管制体系
泰国对现代药品实行四类管制体系(依据《药品法》),药品类别决定销售渠道与操作要求:
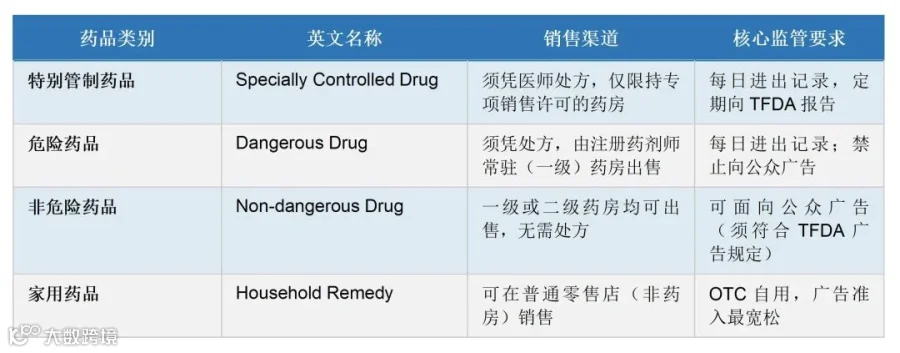
注:泰国【危险药品】(Dangerous Drug)对应实质上的处方药,其范围较宽,包含抗生素、抗高血压药、抗组胺药、疫苗等77.4%的已注册药品;不可简单类比中国的「甲类OTC」理解。须注意,"危险药品"(Dangerous Drug)类别中,并非所有品种均强制要求凭处方才能购买;部分危险药品可在注册药剂师在场督导的一级药房中直接购买,无需处方。仅"特别管制药品"(Specially Controlled Drugs)在法律层面强制凭医师处方购买。这是泰国药品分类体系与中国处方药/OTC分类制度的一个重要实质性差异,须在营销策略和分销渠道规划中予以注意。
4.2 药房分级制度
泰国药房分为两类,决定可经营药品的范围:
● 一级药房(Type 1 Modern Community Pharmacy):持牌注册药剂师须全程在场,可经营所有类别现代药品,包含特别管制药品和危险药品;
● 二级药店(Type 2 Drugstore):由经认证的非药剂师经营,仅可销售有限的非处方类药品。自2005年起TFDA已停止审批新增二级药店执照。
4.3 特殊管制物质的附加要求
若出口产品含有麻醉品或精神活性物质,除正常药品注册外,进口须另行向TFDA麻醉品管制司申请专项进口许可,并严格遵守储存、记录和盘点规定,相关程序更为复杂,须提前规划。

药品标签与包装合规要求
5.1 法定必须标注的信息
根据《药品法》及TFDA相关规定,药品标签须包含以下法定强制要素:
● 产品名称(含通用名);
● TFDA注册号(Drug Registration Number);
● 许可持有人(进口商/分销商)名称及在泰地址;
● 制造商名称及地址;
● 活性成分及含量;
● 剂型与规格;
● 用法用量;
● 储存条件;
● 批号、生产日期与有效期;
● 净含量/数量;
● 适应症/用途;
● 警示语(依据药品类别,危险药品须标注处方药警示)。
5.2 语言要求
泰国对药品标签的语言有明确要求:根据药品类别和使用对象,标签语言要求有所不同:面向普通消费者(家用/非处方类)的标签须以泰文为主;专业医疗机构使用的部分产品可接受英文或泰英双语。但所有法定强制信息(注册号、许可持有人等)需符合TFDA审核的泰文版本。字体高度、最小可读性等有具体要求。
标签须在注册时经TFDA审核批准,未经批准不得擅自更改。建议在产品注册阶段同步准备符合TFDA要求的泰文标签样本,并纳入注册申请材料。
5.3 变更申报要求
如标签内容(含成分信息、规格、储存条件等)发生变更,须事先向TFDA申报变更(Variation Application),经批准后方可执行。中国制药企业生产工艺、包装材料、原料来源或生产地址的变更,均须通知本地许可持有人,由其启动变更申报程序。

中泰两国监管体系关键差异对照

实务建议
7.1 合作协议须明确约定关键条款
中国制药企业与泰国本地合作方之间的协议不应停留于「代理注册」层面,建议明确约定:
● 进口商许可证(Importer's License)的持有与年度续期责任,须确认合作方具备合法进口资质;
● 上市许可的有效期管理(7年期)及续期操作责任;年度许可证维持费缴纳分工;
● LPI申请的操作责任与时效安排,以及因LPI审批延误导致货物滞港的风险分担;
● 变更申报配合义务:中国制药企业变更生产工艺、包装材料、原料来源、生产地址等须提前书面告知并协助申报(Variation Application);
● 本地授权代表(Local Authorized Representative)的委任及其履职要求;
● 知识产权保护条款,防止注册信息被滥用;
● CPP/FSC文件的出具与大使馆认证费用分担安排。
7.2 提前规划LPI申请节奏
依据2025年5月31日生效的新规(FDA通知B.E. 2568),LPI须在货物抵达前通过THAI NSW系统提交,实操中通常在货物预计抵达前2至3个工作日提交申请即可,TFDA审批周期一般为1至2个工作日。但考虑到THAI NSW系统操作学习曲线、HS编码分类核实、首次申报的系统注册等前置步骤,建议在首次发货时预留更充裕时间(如2至4周),待操作熟练后可将节奏压缩至货物抵达前5至7个工作日完成申报。
7.3 标签合规优先处理
泰文标签不符合要求是常见的清关障碍。建议在产品注册申请阶段同步完成泰文标签设计与TFDA审核,确认:所有法定信息完整、语言符合要求(泰文为主)、注册号标注正确、危险药品处方药警示语已依法标注。注册批准的标签须严格执行,任何变更须启动变更申报,不可擅自更改。
7.4 CPP/FSC大使馆认证须提前安排
与许多东南亚市场不同,泰国要求提交注册申请时,中国出具的CPP及FSC须经泰国驻华大使馆认证(Legalization)。大使馆认证流程通常需要数周,须纳入整体注册时间线统筹规划,避免因认证文件未及时取得导致注册申请延误。
7.5 年度许可证管理机制化
泰国进口商许可证、销售商许可证均须每年(1月1日至12月31日)申请续期,逾期将导致许可失效,直接影响进口和销售业务的合法性。建议建立许可证到期前提醒机制,将年度续期列入合作方协议的明确义务,并设定相应违约责任条款。

结语
泰国的药品监管体系以《药品法》B.E. 2510为骨架,以中央集权式管理为特色,兼具全程许可管控和逐批通关机制。从药品注册(Marketing Authorization,7年有效期),到进口商许可证(1年期,须年度续期)、逐批LPI(通过THAI NSW系统)、生物制品批放行,再到清关申报(含大使馆认证文件要求)、GDP仓储与分销,到泰文标签合规,每个环节均有独立的法规要求,任何一个环节的疏漏都可能导致进口受阻或市场退出风险。
中国制药企业进入泰国市场,尤其须重视以下三点有别于其他东南亚市场的特殊要求:CPP/FSC须经大使馆认证、进口商许可须年度续期、逐批LPI须通过THAI NSW电子系统提交并与海关联网。
遵循本指南框架,结合具有泰国药政经验的专业顾问的持续合作,将有效降低合规风险,为中国药品顺利进入泰国市场奠定坚实基础。



声明:①本公众号内容仅用于信息交流,素材均来源于公开渠道。如您认为相关内容涉嫌侵权,请通过微信:wxid_77ngiuv2gdzq22 提交书面通知(请注明“侵权投诉”),我们将在24小时内核查并依法处理。②商务合作及内容转载授权,请联系上述微信(添加时请备注事由)。