点击上方蓝字关注我们


01
-
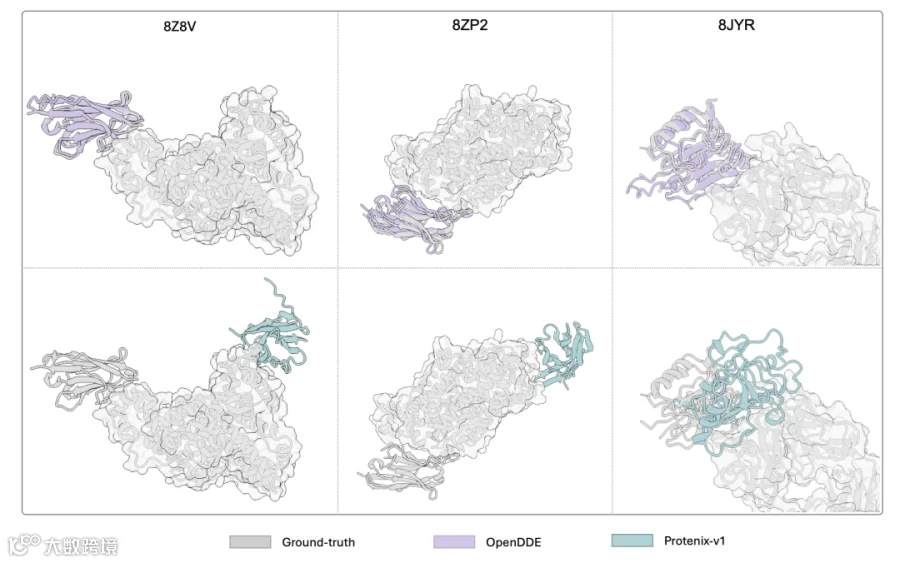
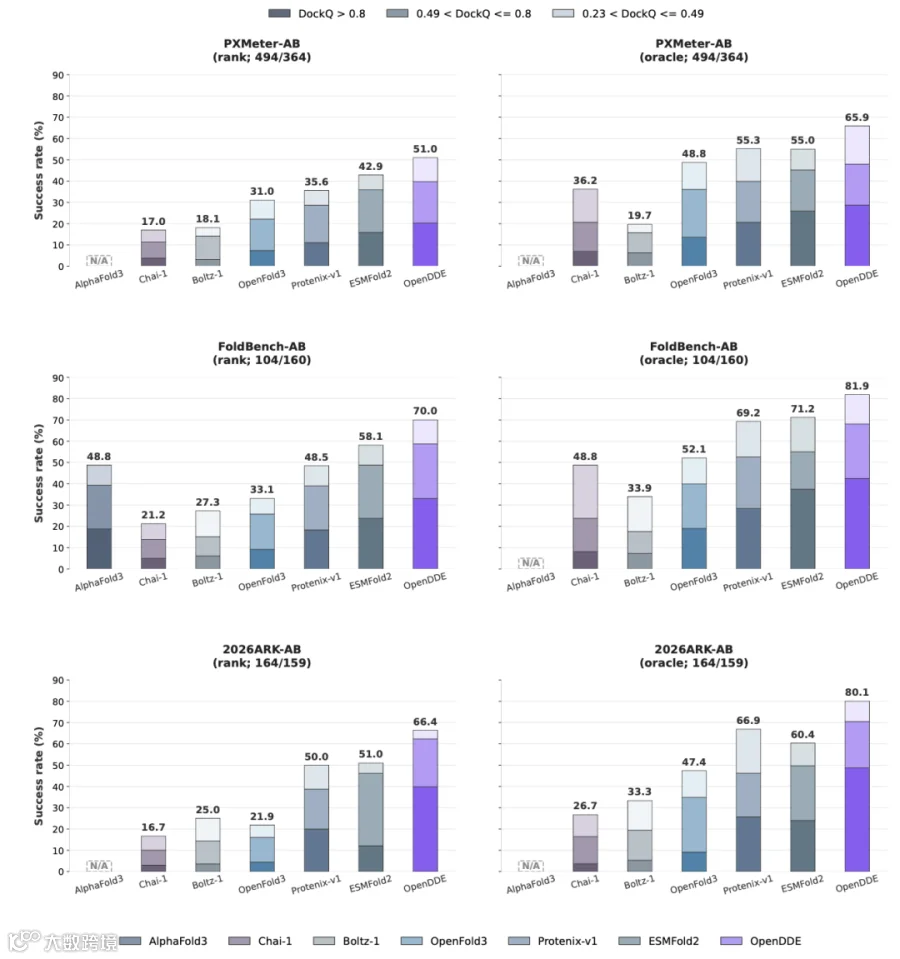
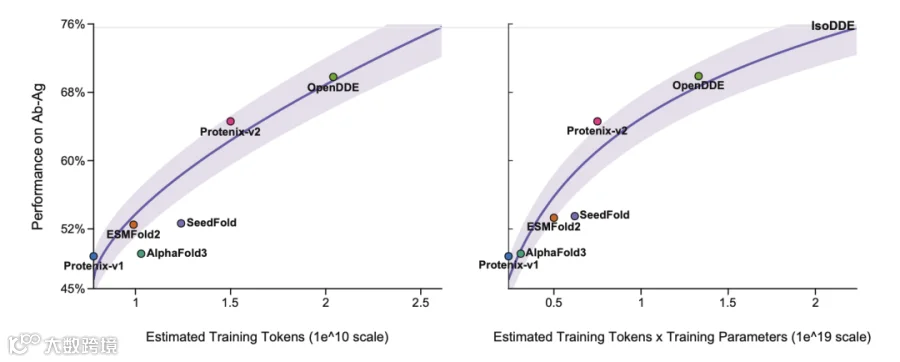
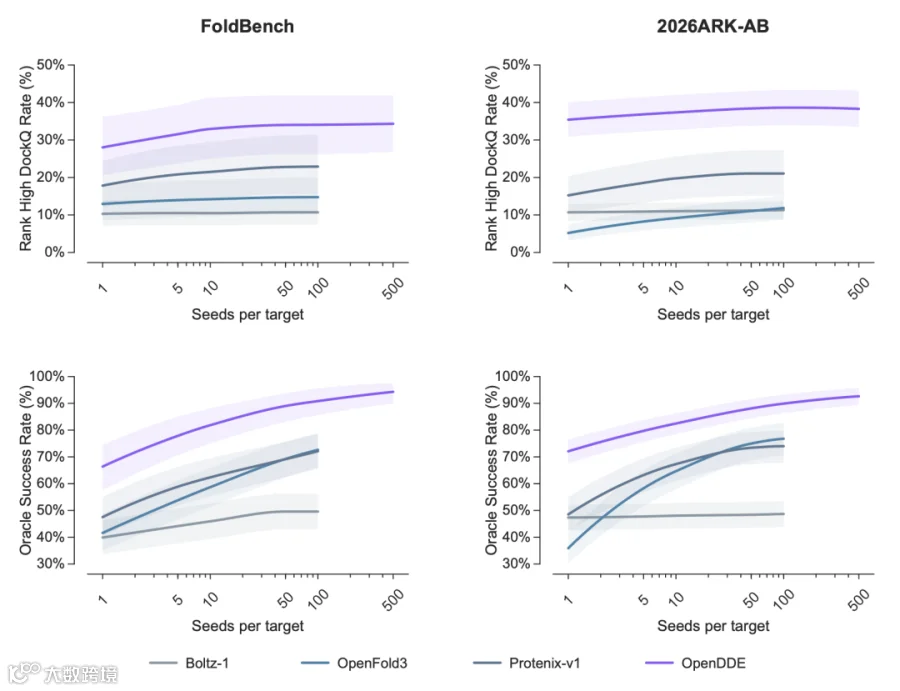
PXMeter-AB : OpenDDE 以 51.0% 的成功率( DockQ > 0.23 )登顶,而 ESMFold2 为 42.9% , Protenix-v1 仅 36.2% , Chai-1 更是只有 17.0% 。
-
FoldBench-AB : OpenDDE 达到 70.0% ,将 ESMFold2 的 48.8% 和 AlphaFold3 的 48.8% 远远甩在身后。
-
2026ARK-AB (全新发布的时间外验证集): OpenDDE 以 66.4% 的成功率大幅领先 ESMFold2 的 51.0% 、 Protenix-v1 的 50.0% ,以及 Chai-1 的 16.7% 。
02
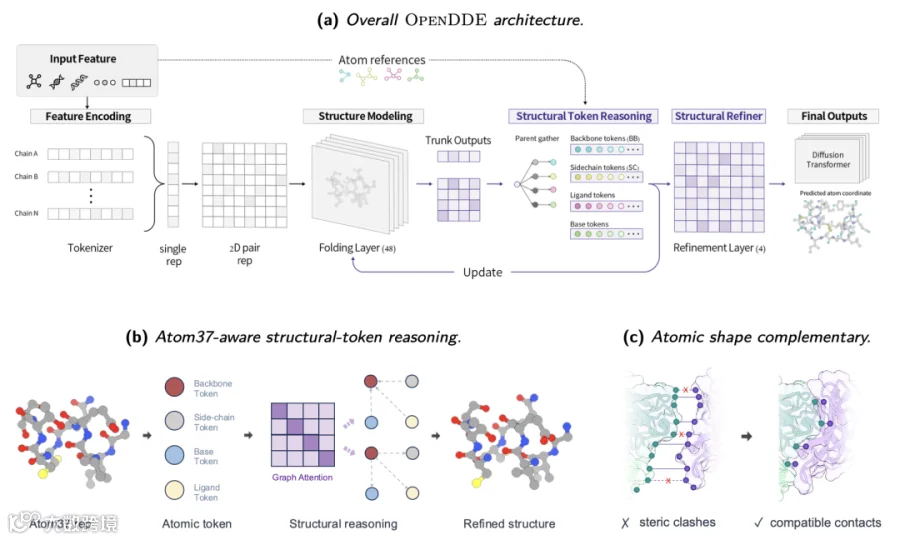
残基 token → 结构 token → 原子坐标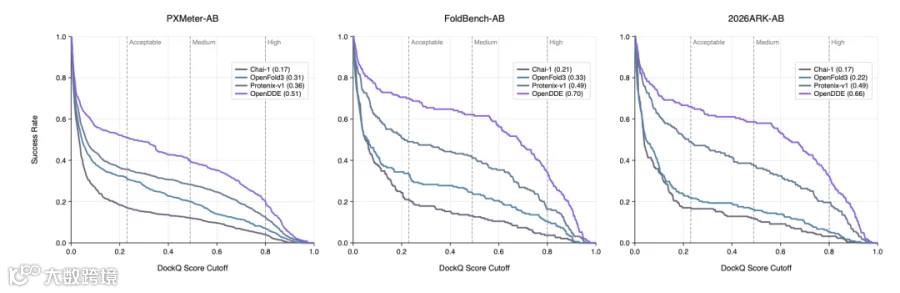

从头设计 = 部分原子作为已知条件固定,模型只去噪未知部分
03
04