
2026-03-12
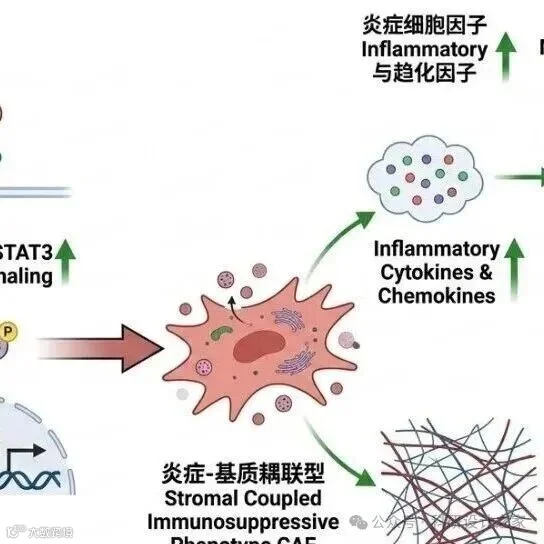
2026-03-12

2026-03-11
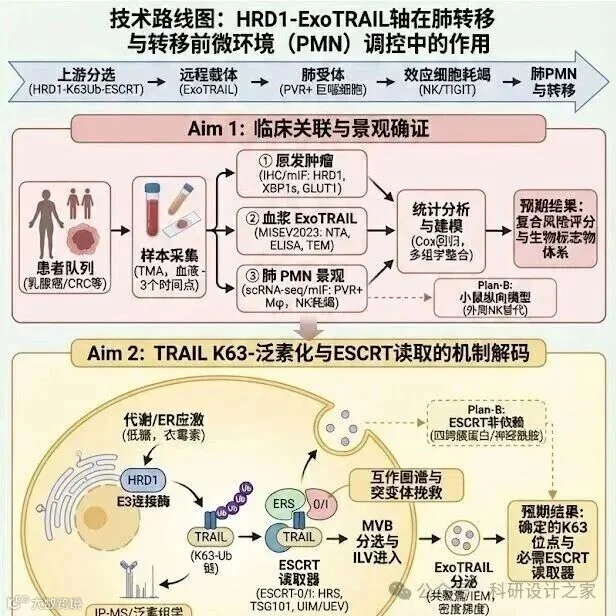
2026-03-11
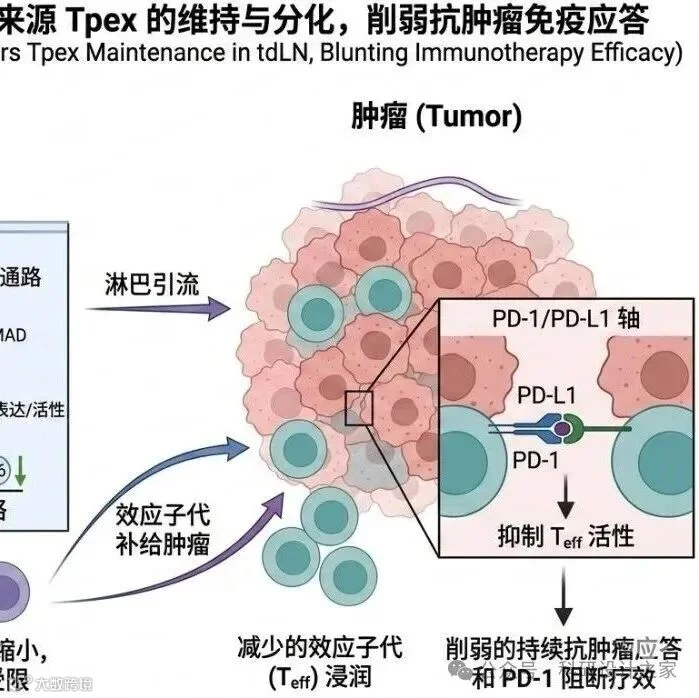
2026-03-08
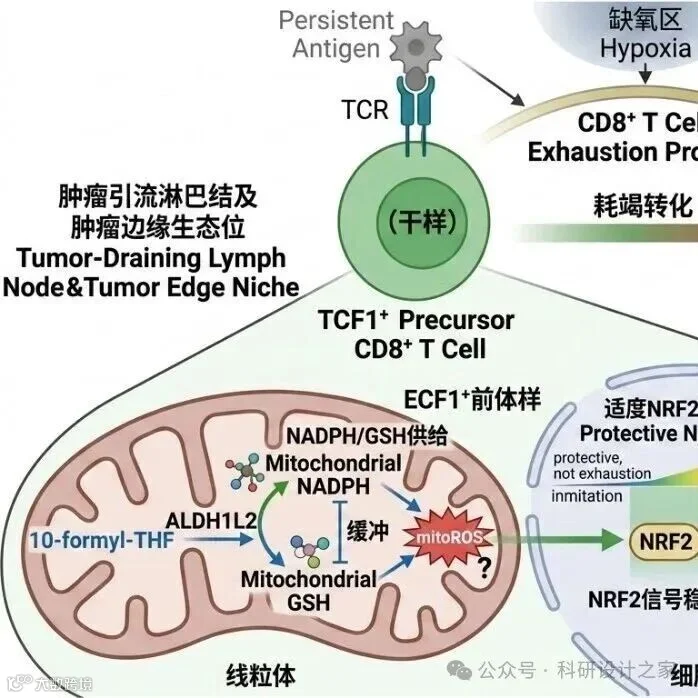
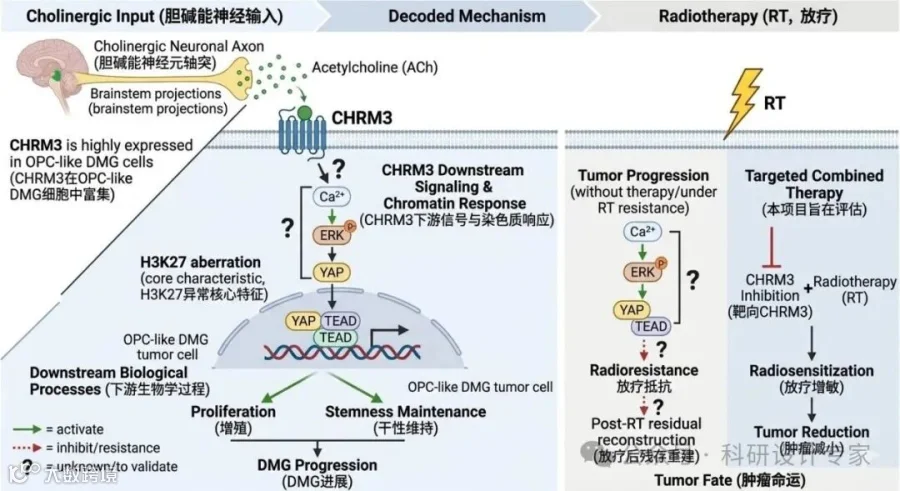
适配2026年国自然青年C类项目逻辑框架(模板初稿)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

 生物医学AI圈子
生物医学AI圈子
2026-03-12
2026-03-12
2026-03-11
2026-03-11
2026-03-08
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|