


引用本文
王欣,赵余,李珍珍,王进涛,许建华,李天泉*.激活市场竞争的关键突破口: 简化非处方药上市流程[J].中国食品药品监管,2026(1):100-107.
激活市场竞争的关键突破口: 简化非处方药上市流程
Unlocking Market Competition: Simplified Market Access Pathways for Over-the-Counter Drugs
王欣
重庆康洲大数据(集团)有限公司
WANG Xin
Chongqing Kangzhou Big Data (Group) Co., Ltd.
赵余
重庆康洲大数据(集团)有限公司
ZHAO Yu
Chongqing Kangzhou Big Data (Group) Co., Ltd.
李珍珍
重庆康洲大数据(集团)有限公司
LI Zhen-zhen
Chongqing Kangzhou Big Data (Group) Co., Ltd.
王进涛
重庆康洲大数据(集团)有限公司
WANG Jin-tao
Chongqing Kangzhou Big Data (Group) Co., Ltd.
许建华
重庆康洲大数据(集团)有限公司
XU Jian-hua
Chongqing Kangzhou Big Data (Group) Co., Ltd.
李天泉*
重庆康洲大数据(集团)有限公司
LI Tian-quan*
Chongqing Kangzhou Big Data (Group) Co., Ltd.
摘 要 / Abstract
在我国部分化学药非处方药(OTC)领域,因缺乏有效竞争,消费者的选择受到限制,其核心症结在于无参比制剂的化学药OTC 药品上市审批难度较大。现行法规要求证明仿制药与原研药品的生物等效性,但对于维生素、矿物质等历史品种,参比制剂缺失以及传统临床试验难以实施等因素,造成企业研发路径不明、成本高昂,部分新竞争者难以进入市场。本文剖析了我国针对无参比制剂化学药OTC 药品的现行法规要求和国际OTC 药品管理的成功经验,提出了构建契合我国国情的分级分类化学药OTC 管理体系改革相关思考:建立简易申请OTC 目录,针对高安全性品种豁免参比制剂要求,简化注册流程;优化化学药OTC 审评审批路径,明确评价标准;建立目录动态调整与持续监管机制。
In certain segments of over-the-counter (OTC) chemical drugs in China, a lack of effective competition has limited consumer choice. The core issue lies in the high barriers to market authorization for OTC chemical drugs that lack reference listed drugs (RLDs). Current regulations require generic drugs to demonstrate bioequivalence with originator products. However, for historically established categories such as vitamins and minerals, the absence of RLDs and the impracticality of conducting traditional clinical trials have led to unclear research and development pathways, high development costs for enterprises, and significant barriers for new market participants. This paper analyzes China’s current regulatory requirements for OTC chemical drugs without RLDs and draws on successful international practices in OTC drug regulation. It further proposes reform ideas for establishing a tiered and categorized management system for OTC chemical drugs suited to China's national context. Key recommendations include establishing a simplified application catalog for OTC drugs; exempting high-safety products from RLD requirements and streamlining their registration procedures; Optimizing the review and approval pathways for standard OTC drugs while clarifying evaluation criteria; and developing mechanisms for dynamic catalog adjustment and continuous postmarket supervision.
关 键 词 / Key words
非处方药;上市审批;专论制度;参比制剂;分类管理;监管改革
over-the-counter (OTC) drugs; marketing authorization; monograph system; reference listed drugs; categorized management; regulatory reform

在北京市市场监督管理局2023 年开具的一张行政处罚决定书中显示[1],北京朗迪制药有限公司生产的32 批次碳酸钙D3产品,其维生素D3 含量不符合规定,涉及已售出产品93 万余盒。该公司被处以1.34 亿余元罚款,并被责令停产停业整顿。然而,即便历经上述处罚事件,“朗迪钙”系列产品在2023 年的年销售额仍超过20 亿元,在补钙制剂市场中占据领先地位。能取得这样的成绩,很大程度上是因为“朗迪钙”系列产品作为独家品种,缺乏直接竞争品。消费者即便对产品质量存疑,也缺乏更多可供选择的替代品。这一现象背后,折射出我国化学药非处方药(over-the-counter drug,OTC)市场的深层次问题,即部分领域竞争不充分,消费者选择空间受限。这种市场失衡的部分根源在于无参比制剂的OTC 难以获批上市的困境。当潜在竞争者难以进入市场时,已有品种持有人自然缺乏提升产品的内在动力。

01
审批困境:无参比制剂化学药OTC 上市面临的难题
处方药与OTC 在临床使用、风险程度和公众可及性等方面存在本质区别。处方药是指必须凭执业医师或执业助理医师处方,方可购买、调配和使用的药品。此类药品具有药理作用明显、所治疗病情较重、使用风险较高等特点,须在医师指导下使用,其用药决策依赖专业判断。与之相对,OTC 是指无需医师处方,消费者可自行判断、购买和使用的药品。其核心特征在于有效性明确,安全性范围广,误用和滥用风险较低,适应症和用法用量确定,且具备便于消费者自行判断、使用与管理等特点。此外,OTC在剂型、规格、口味、包装等方面的设计,更贴合消费者的个性化需求。
基于上述差异, 两类药品的监管思路理应适应各自特性。但在现行化学仿制药审评体系中,这一原则还尚未充分体现。2020 年发布的《药品注册管理办法》[2] 和《化学药品注册分类及申报资料要求》[3] 明确,化学药品3 类、4 类和5.2 类均要求证明与参比制剂的质量和疗效一致。2023 年发布的《国家药监局关于无参比制剂品种仿制研究的公告》及政策解读[4] 中明确,对于无参比制剂品种的仿制研究,要坚持以临床价值为导向,坚持高标准、严要求。这类药品首先应解决是否具有临床价值问题,也就是通过临床试验获得完整充分的安全性有效性数据,证明其临床价值。以上法规和政策未区分处方药和OTC。然而,部分化学药OTC上市时间较早,存在参比制剂不明确、无参比制剂的情况。理论上,无参比制剂药物可以通过临床试验证明其有效性后申请上市,但是像维生素、碳酸钙等部分品种,较难通过临床试验来证实其有效性。部分企业在综合风险、成本与收益后,往往只能选择放弃相关品种的研发。
国家药品监督管理局(以下简称国家药监局)药品审评中心2020 年发布的《化学药品非处方药上市注册技术指导原则(征求意见稿)》[ 以下简称《指导原则(征求意见稿)》][5] 中提出:“(一)认可在已上市的非处方药基础上,结合适应症和品种特点,开发适合我国人群喜好及用药习惯且质量可控的非处方药品种。(二)非处方药临床价值(临床需求)的评估,以满足我国人群的个性化用药需求为考虑,不做优势性比较。(三)长期广泛的境内外人用经验可以作为重要证据,用于评估非处方药在我国上市的获益风险,并根据评估结果,考虑适当简化或豁免上市注册相关临床研究。”《指导原则(征求意见稿)》对化学药OTC 产品上市有诸多利好,例如:对改剂型、改规格、改复方的化学药OTC 新药,不要求其具备临床优势,而化学药处方药的改良型新药需要具备临床优势才能上市;对于已有充分证据支持安全有效的OTC 品种,可以利用现有数据简化或豁免临床研究;对境外已上市的OTC,可基于其上市基础与我国指导原则,酌情减免临床研究要求。然而,遗憾的是,其至今尚未发布正式文件。即便《指导原则(征求意见稿)》最终得以落地实施,对于无参比制剂的化学药OTC 而言,其上市之路依然充满坎坷。《指导原则(征求意见稿)》虽提及“对于难以明确参比制剂的非处方药品种,在认可临床价值(临床需求)的前提下,可采用质量提升方式开展仿制药评价”。但部分环节仍缺乏明确、可操作的定义和评价体系,例如,“临床价值”如何进行评价与认可,是通过验证性临床试验、依托人用经验数据,还是理论推导即可,还需进一步明确;“质量提升”具体指什么,需要达到何种标准,与何种对象进行比较方能认定为“提升”等。这些问题使得企业在开展研发工作时如同在黑暗中摸索,监管部门在审评过程中也缺乏统一尺度,此类申请易陷入僵局。
2020 年,国家药监局药品审评中心还发布了《临床价值明确,无法推荐参比制剂的化学药品目录(第一批)(征求意见稿)》[6]。该目录涵盖氯化钠注射液、葡萄糖注射液、维生素B2 片等117 个临床价值明确但无国际参比制剂的品种。但目前该目录尚未发布正式文件。2023 年10 月,《国家药监局关于无参比制剂品种仿制研究的公告》[4] 发布,明确无参比制剂品种需要自证临床价值,且境内外已有公开数据,以及之前公示的《临床价值明确,无法推荐参比制剂的化学药品目录》中所说的“临床价值明确”,不能作为自证临床价值的证据。企业需要自行通过良好设计的临床试验证明相关品种的预期临床价值。
在严苛要求和高昂成本的双重作用下,市场形成了一个“死水效应”:独家产品因缺乏竞争而高枕无忧(即使有质量问题),而新进入者则因路径不明、成本过高,只能望而却步,最终导致部分细分领域长期处于缺乏有效竞争的状态。

02
国际借鉴:OTC 专论制度
在审视全球药品监管实践时, 美国的OTC 专论制度[7](OTC Monograph) 提供了一个极具参考价值的模式。该制度的核心在于, 由美国食品药品监督管理局(Food and Drug Administration,FDA) 组织专家对特定类别的OTC 药品成分、剂量、剂型、适应症和标签进行全面、公开的科学评估,形成具有法律效力的“专论”。任何生产商只要确保其产品符合相应专论的标准(包括成分、规格、标签、生产质量等),即被视为安全有效(generally recognized as safe and effective,GRASE),对符合暂定最终专论或最终专论条件的OTC, 仅需进行生产前登记, 提供标签、配方信息、安全性信息等资料, 符合《现行良好生产规范》(Current Good Manufacturing Practices,cGMP)生产标准,取得美国国家药品代码(the National Drug Code,NDC)后即可生产销售,无需进行等效性评价、临床试验,也无需再经过FDA 的逐一审批。
这一制度的构建经过了严谨的科学评估流程:组建专家顾问审查组- 全面评估数据- 分类决策(安全有效/ 非安全有效/ 数据不足)- 多轮公示与修订。2020年3 月,美国进一步通过《冠状病毒援助、救济和经济安全法》[8](Coronavirus Aid, Relief, and Economic Security Act,CARES Act),该法案在《联邦食品药品和化妆品法案》(Federal Food, Drug and Cosmetic Act) 中增加了第505G 条。第505G 条对OTC 专论药品的监管框架进行了改革与现代化升级,建立了更高效的行政命令程序:允许行业主动发起OTC 专论命令请求(OTC Monograph Order Request,OMOR),FDA 接受后发布建议命令,经45 天以上公众评论期,最终命令经争议解决程序后正式生效。这一规定适用于拟发起专论修订的所有OTC 产品。
上述OTC 专论制度在实践中体现为如图1 所示的美国OTC仿制药上市路径:当仿制药活性成分已纳入OTC 专论时,若完全符合专论标准则仅需企业登记备案即可上市;若不符合则需发起OMOR,经公众评论后修订专论。如果活性成分未列入OTC 专论,则必须通过简略新药申请(abbreviated new drug application,ANDA)提交完整材料证明与原研药一致,经FDA审评批准后方可上市。
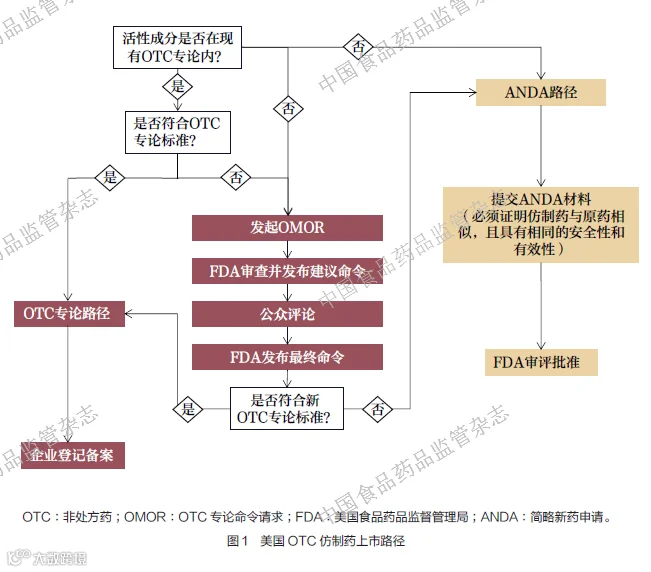
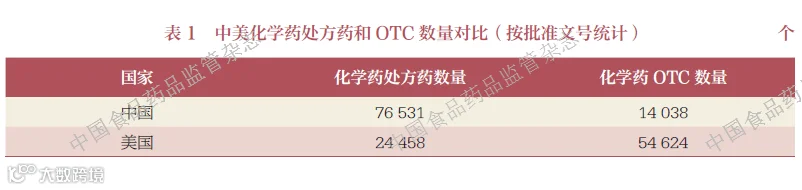
这一制度极大促进了美国OTC 市场的良性竞争。制药企业无需投入巨额资金进行重复研究,只需确保产品符合既定专论标准即可上市销售,显著降低了市场准入门槛。所以,在美国,消费者可以选择的OTC 产品更加丰富。截至2025 年6 月25日,按批准文号统计,美国上市的化学药OTC 数量为5.4 万多个,化学药处方药数量大约只有OTC 的一半;而我国上市的化学药OTC 数量为1.4 万多个, 不到化学药处方药数量的20%,见表1。大约有1200 余个OTC 活性成分(多数为复方成分)在美国上市而未在我国上市。
日本市场同样设有OTC 简化上市机制, 即OTC 许可标准制度。1969 年日本厚生劳动省(Ministry of Health, Labour and Welfare,MHLW) 启动OTC 许可标准建立工作,旨在提升OTC 产品审评效率,推动审评审批程序合理化和透明化。1970~2017 年, 日本已针对16类OTC 以及一些类别的医药部外品建立了国家生产销售许可标准[9],许可标准包括范围和标准的具体内容两大部分,其中具体内容又分为活性成分的种类、活性成分的用量和配方规则、剂型、用法用量、适应症、包装单位6 个方面。对于已颁布正式许可标准的OTC,除部分药品外,注册申请均授权地方政府实施批准,其他类别的OTC 申请则由日本药品及医疗器械综合机构(Pharmaceuticals and Medical Devices Agency,PMDA)审评,MHLW 批准。日本OTC 许可标准制度减轻了制药企业开发负担,加快了OTC 产品上市进程,经授权地方政府负责的OTC 药品注册申请流程,相较于未授权地方政府负责的流程,审批时间可缩短7~8 个月[9]。截至2025 年6 月25 日,日本市场上大约有1760 余个已上市OTC 活性成分(其中多数为复方成分)品种尚未在我国上市。

03
监管思考:建立契合我国国情的化学药OTC分类管理体系
中国[5]、日本[9] 和美国[10] 均将OTC 界定为公众可自行识别、选择并安全使用,且无需专业医疗干预的药品,这一共识决定了“以风险分级为前提、以科学评价为基础、以简化注册为手段”的OTC 监管理念具有跨市场的通用性。FDA 与MHLW 在建立OTC 专论或许可标准制度时,均先组织多学科专家对已上市产品开展了系统性评价,形成“公开透明、可验证、可复现”的科学结论,再以法规形式固化[7,9]。该路径既保证了审评结论基于循证数据,也为后续企业“备案即可上市”提供了可预期的技术标准。
基于国际经验和国内实际,笔者提出建立分级分类的OTC管理体系等系统性思考,旨在为OTC 产品开辟科学的上市路径。
3.1 为高安全性化学药OTC 建立“ 简易申请OTC目录”
组织由临床医学专家、药学专家、药品监管专家等组成的权威专家组,基于国内外药品不良反应监测数据和丰富的临床使用经验, 筛选出不良反应发生率低且长期使用安全性高的化学药OTC 品种,形成“简易申请OTC 目录”并分批发布,从试点执行到逐步扩大(例如,国内长期使用且未出现严重不良反应的矿物质类、维生素类药品等,可作为首批优先纳入目录的品种)。对于纳入目录清单的产品,豁免与参比制剂的对比要求。申请人需向国家药监局重点提交以下资料:药品的质量标准、生产工艺描述、稳定性研究资料,以及安全性和有效性评价资料。其中,安全性和有效性评价资料可参考国内外已有的研究数据和临床使用经验。这些资料将作为审评审批的核心依据,确保在简化审评审批流程的同时,依然能够保障药品的质量和安全性。
3.2 优化化学药OTC 的上市路径
对于其他化学药OTC 产品,特别是目前仍按甲类管理的化学药OTC,可在现行法规与技术审评框架内,通过制度细节的再设计实现渐进式优化。例如,加速《化学药品非处方药上市注册技术指导原则》正式文件的发布,推动“对于难以明确参比制剂的非处方药品种,在认可临床价值(临床需求)的前提下,可采用质量提升方式开展仿制药评价”的政策条款落地。具体而言,药品监管部门应在配套问答、模板案例及培训材料中,逐步细化“临床价值”的内涵,评价体系可以参考《药品临床综合评价管理指南(试行)》,但需要结合OTC 的“自我药疗”特征,在指标权重上可考虑向“安全性”“可及性”“误用/ 滥用风险可控性”倾斜;同时,针对“质量提升”制定包含原料药晶型控制、杂质谱一致性、体外溶出曲线对比、稳定性数据包等在内的技术评价标准体系,并允许企业以补充研究、分阶段滚动提交的方式完成证据链闭环。通过上述举措,既保留原有审评体系的连贯性,又为OTC 研发生产企业提供一条可操作、可预期、可落地的注册路径,最终提升我国化学药OTC 整体质量水平与市场供给效率。
3.3 建立动态调整与持续监管机制
自上而下建立多维度、全流程的公众反馈渠道,广泛听取医疗机构、药品研发生产企业、行业协会和消费者等的意见和建议。对于收到的反馈信息,按安全性、有效性、可及性三维度分类,组织临床医学、药物流行病学、质量管理等领域专家进行分析论证。分批发布或及时更新“简易申请OTC 目录”,以确保目录的科学性和实用性。同时,依托国家药品不良反应监测系统,对目录内品种实施主动监测与自发报告“双轨并行”,重点聚焦儿童、孕产妇、老年人等特殊人群用药风险;制定年度质量抽检计划,覆盖生产、流通、使用等全链条,且抽检结果实时向社会公开。一旦确认某品种存在安全性隐患或质量波动,立即启动目录退出程序:先行发布风险警示,7 个工作日内完成企业问询与现场核查,确认后48小时内发布除名公告,并同步向医保支付、招标采购、零售药店等各相关环节通报,确保风险药品在最短时间内退出市场,最大限度保障公众用药安全。
当然,任何制度改革都伴随挑战。在推行“简易申请OTC目录”与优化审评审批路径的过程中,也需警惕可能出现的新问题:一是简化审评审批流程是否会导致低水平重复申报,造成资源浪费与市场拥挤;二是地方监管部门能否有效应对分级分类审评审批所带来的监管范围扩大与监管压力倍增的双重挑战;三是如何在鼓励创新的同时,守住药品安全底线,避免“重审批、轻监管”。这些问题的解决,有赖于配套制度的同步完善,如强化企业主体责任、建立全国统一的OTC 药品追溯系统、实施动态目录调整与退出机制等,从而形成“放得开、管得住”的良性治理格局。

04
展望与结语
当下,国内OTC 销售渠道正迎来变化:广东省推动乙类OTC 跨界融合改革,试点在大型连锁便利店内经营乙类OTC[11] ;四川省促进药品流通产业高质量发展,支持药品零售(连锁)企业设置专柜或自动售药机销售乙类OTC[12] ;O2O 模式实现即时送药,30 分钟即可送达消费者手中;医保电子支付实现线上线下全渠道贯通……多元化的消费场景已基本打通了药品销售“最后一公里”的堵点。当下,若OTC审评审批制度能够有所革新,加速更多安全有效的OTC 产品上市,将形成供给扩容与渠道释放的“双轮驱动”格局。届时,我国OTC 市场将从“ 价格竞争”转向“品质与服务竞争”,消费者在居住地周边即可购买到与国际同步上市的新品好药。行业整体规模有望持续扩大,并带动上下游的研发、生产、流通、零售全链条升级,真正让“双轮驱动”成为推动健康中国建设的新引擎。

第一作者简介
王欣,硕士,重庆康洲大数据(集团)有限公司。专业方向:医药市场政策研究
通讯作者简介
李天泉,硕士,重庆康洲大数据(集团)有限公司,正高级工程师。专业方向:医药市场政策研究
