

PDA TR 60《工艺验证:生命周期方法》(以下简称技术报告)是制药行业工艺验证领域的重要技术文件。近期,该技术报告修订了2026新版本。
为此,我们在6月3日晚上安排了一场免费直播,由专家深度解析本次修订要点。目前活动已经顺利结束,感谢大家的支持与参与!我们同步整理了课程内容及答疑(见文末),以供参考。
本次修订要点主要有以下三个方面:
1.工艺参数(PP)关键性决策树的优化
2.工艺性能确认(PPQ)批次数的确定和取样策略
3.分阶段持续工艺确认(CPV)的方法
工艺参数关键决策树的优化
两个版本的工艺参数分类对比:
|
2013版 |
2026版 |
|
关键Critical:影响产品质量 非重要Non-key:参数本身容易控制,或有着很宽泛的接受限度。如果可接受限度超出,非重要工艺参数可能会对质量或工艺性能产生影响。 |
关键Critical 非关键Noncritical
非关键工艺参数:不会对该参数所涉及的工序的质量。但可能会对下一个工序的工艺性能有影响,从而需要监控。 |
相关术语:
关键工艺参数(Critical Process Parameter,CPP)
非关键工艺参数(Non-Critical Process Parameter,Non-CPP)
重要工艺参数(Key Process Parameter,KPP)
非重要工艺参数(Non-Key Process Parameter,NKPP)
旧版当时存在的问题:非重要工艺参数(NKPP)中对于“宽泛的接受限度”,在不同部门之间理解不一致。
新版主要做了以下三方面的调整:
1.在参数分类定义上明确区分“产品质量”和“工艺性能”
产品质量:最终药品是否安全、有效
工艺性能:生产过程是否顺畅、稳定、高效
2.弱化NKPP的概念
新版调整了NKPP的命名,将参数分为:关键、非关键(重要、操作)。其中“操作”对当前工序质量无影响,但可能影响下一工序的工艺性能,因此需要监控。
3.明确KPP不是法规强制要求
新版明确说明KPP的识别不属于法规强制要求。其他的监管机构的立场:
国家药监局药品审评中心(CFDI)发布的 2025版《工艺验证检查指南》中没有要求企业必须识别KPP。
FDA 2011版工艺验证指南同样没有要求企业必须识别KPP。该指南指出:参数的重要性不应简单分为“是/否”两类,而应视为一个连续谱系。在生命周期方法中,基于风险的决策比僵硬的分类更有意义。
专家建议:过度细化非关键参数的评估反而会降低效率。企业应根据自身产品和工艺的实际情况,自主决定是否需要识别KPP。
工艺关键决策树的评估逻辑变化
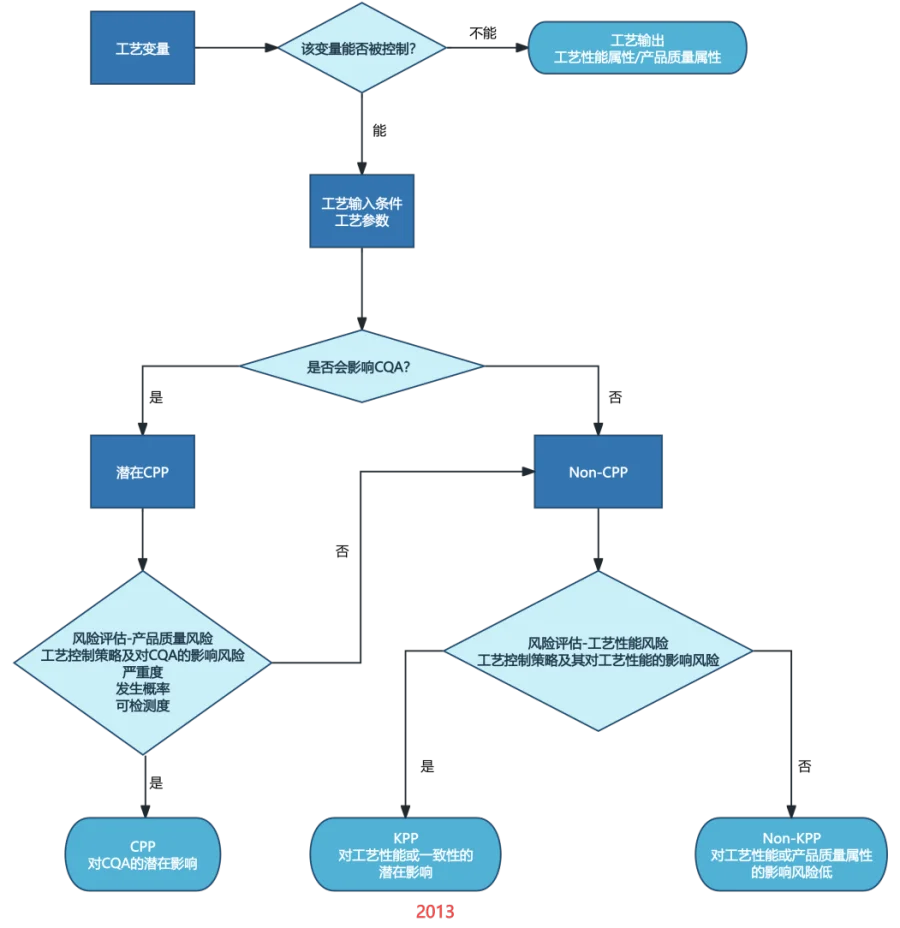
(点击放大查看图片)
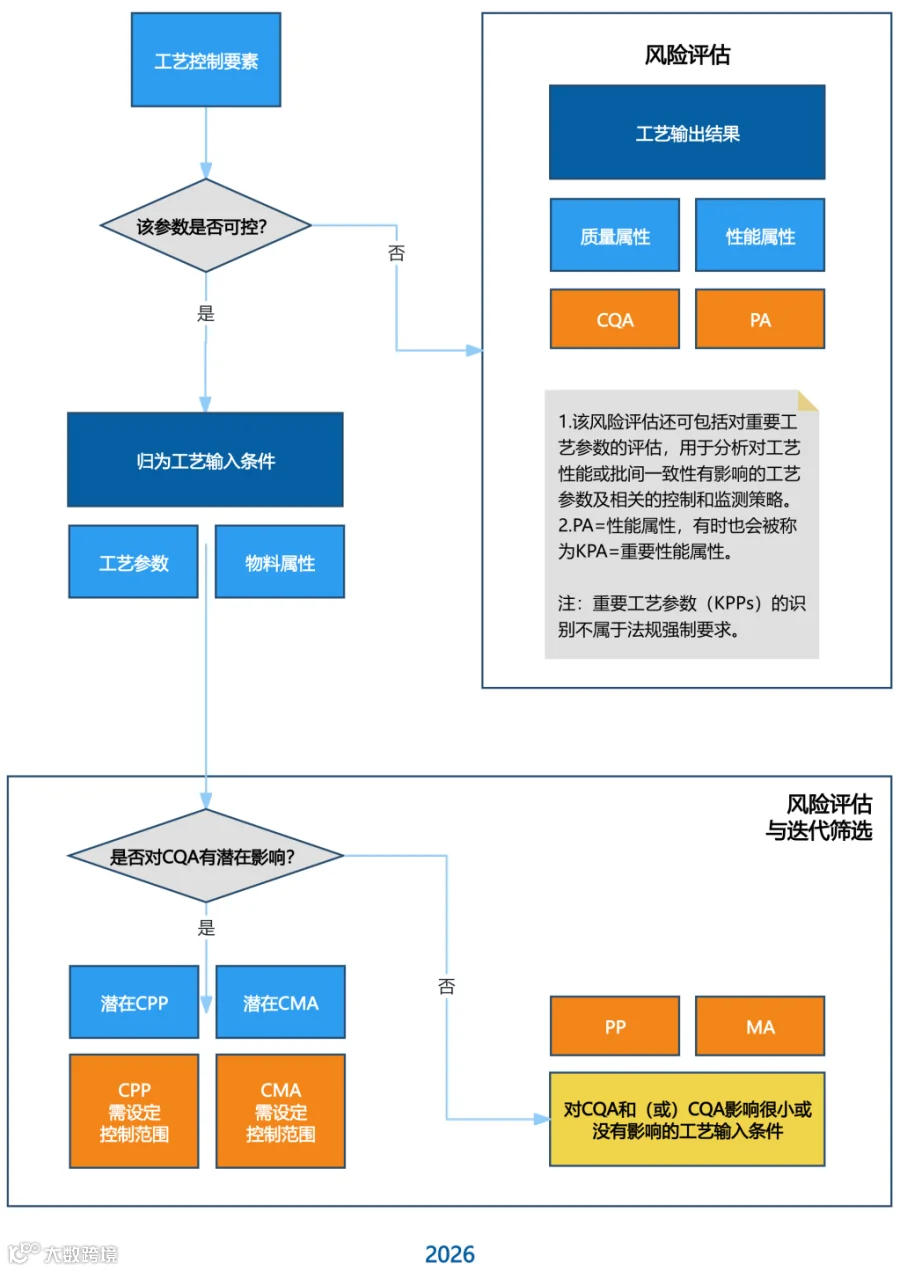
(点击放大查看图片)
工艺性能确认批次数与取样策略
批次数如何确定?
26版引入了“baseline”概念,作为确定批次数的起点。
多数企业会将法规通常要求的“连续3批”理解为baseline(基线);
指南也提出欧盟GMP附录15要求至少3个连续批次可作为baseline制定的基础。
批次数调整的逻辑:
对工艺了解较少,或工艺本身复杂 → 需要增加批次或加强监测
对工艺了解充分,且工艺本身不复杂 → 经科学论证后,可以少于基线批次
批次数确认的步骤:
1.确定基线(基于产品和工艺知识)
2.评估额外控制(是否需要额外技术论证/活动)
3.评估非GMP风险(如业务、技术、战略影响)
取样策略如何制定?
新版将旧版关于关键质量属性(CQA)的“风险严重性”重新定义为“伤害严重性”。并且,新版在统计评估的例子中增加了“比例(coverage)”的举例,与旧版列举的“置信度”形成互补:
置信度:评估结果的可信程度
比例:预测整体产品落在规格范围内的百分比
|
伤害严重性(Harm Severity) |
置信度/比例 (%) |
|
高严重性(如效价Potency) |
95/99 |
|
中等严重性(如渗透压Osmolality) |
95/95 |
|
低严重性(如pH值) |
95/90 |
取样数量举例:

应用逻辑:风险越高,要求达到的比例就越高,需要取的样品数量也越多。但特定产品剂型(如混合均匀度)还需参考相关监管指南。
对于如何计算确定取样数量,指南认为容忍区间法仍是最常用的工具,要通过临床生产/工程批/技术转移等数据,计算工艺的变异性,从而计算目标取样数量。
对于没有先验数据的,指南提到了至少6个样,前中后个2个样,这样可以确保有足够数据评估批间批内差异。
分阶段持续工艺确认方法
为什么需要分阶段持续工艺确认(CPV)?
完成传统的3批PPQ后,工艺验证并没有真正结束。此时只具备了初步的统计意义,达到了上市门槛,但距离“高度保证工艺稳定”还有距离。
技术报告的附录案例显示,3批PPQ完成后,只能覆盖约50%的批间变异。之后进入CPV,在减少取样的前提下再运行4批,等覆盖了75%的批间变异后,才能切换至常规取样方案。
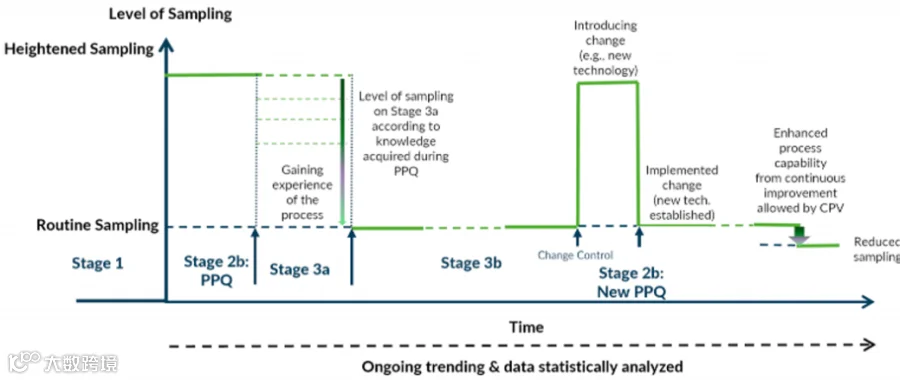
以下图示直观地展示了工艺验证生命周期内不同阶段的取样强度:
(点击放大查看图片)
3a代表更加紧密的取样水平,3b是日常生产的取样水平。
3a:强化监测
核心目的:使用生命周期各阶段数据进行统计评估,及时检测不良工艺变异。
启动时机:PPQ完成后立即启动。
批次数量:基于统计显著性确定(需提供足够数据量以深度理解工艺变异)。指南没有给出批次数建议,可以参考其他信息来源:
ISPE PQLI:建议30批数据计算CpK;
Minitab控制图:建议20个数据点。
取样要求:通常维持PPQ阶段的取样水平,确保数据充分,覆盖所有关键位点。
核心任务:建立工艺目标基线,通过分析PPQ批次的数据,计算出当前工艺的PpK值,作为后续3b日常监测的参考基准,从而确认3b监测计划。
补充说明:
1. ISPE PQLI为国际制药工程协会发布的产品质量生命周期实施指南系列.
2. CpK为过程能力指数,用来衡量工艺在受控状态下的潜在能力:
CpK ≥ 1.33 通常认为工艺能力充足、处于受控状态;
CpK < 1.0 表明工艺能力不足,可能导致超出合格标准。
3. PpK为过程性能指数,用来衡量工艺长期稳定性:
数值越高,表示工艺越稳定、能力越强
数值越低,表示工艺波动越大,容易产出不合格品
3b:常规监测
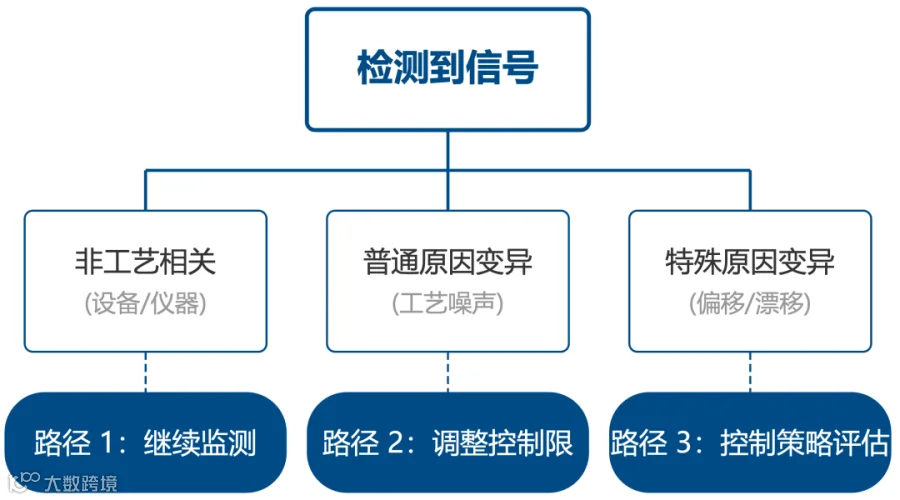
基于信号分类的三条响应路径:
除了上述三大核心修订要点外,新版技术报告还描述了知识管理的策略与案例,以及AI在工艺验证中的潜在应用场景。限于篇幅,这里不再展开,感兴趣的读者可以直接阅读技术报告原文。
结语
2026版PDA TR 60的修订,体现了工艺验证从“一次性事件”向“数据驱动的知识管理体系”的转变。
新版不再要求企业机械套用分类模板,而是鼓励基于科学知识、风险认知和实际数据灵活设计验证策略。
理解这些修订变化的背后的逻辑——为什么要这样分类、为什么要分阶段、为什么重视知识管理,比记忆具体术语更为重要。
直播答疑
以上是本文的全部内容,如果你觉得有帮助,请转发给更多的人~
gempex德恩咨询作为源自德国的GMP咨询与执行机构,深耕合规领域20余年,我们可以提供覆盖产品全生命周期的定制化合规解决方案:
工艺设计阶段:法规解读、差距分析、工艺验证主计划审核、技术转移支持
工艺确认阶段:风险评估、工艺验证方案起草/修订/审核、指导实施、偏差调查与处理
持续工艺确认阶段:工艺验证体系搭建/升级、数据完整性支持、方案报告起草/审核
设施设备、公用系统的确认(IQ/OQ/PQ)
清洁验证支持
我们帮助企业建立验证能力——让合规更高效、落地。点击下方图片,了解我们完整的合规服务类型。

关注“二维码”,或邮件/电话联系我们

服务热线:400 166 2002
邮箱:info-cn@gempex.com

gempex德恩咨询深耕GMP合规领域24年,致力于为全球的生命科学企业提供合规、高效及可执行的GMP解决方案,帮助制药、生物技术、ATMP、原料药、原辅包、医疗器械等各方达到NMPA、EU、FDA、PIC/S等GMP标准,减少合规及药品安全风险。
目前,我们拥有60多位经验丰富的GMP专家,全球累计执行项目超过5000个,累计为1000多个客户提供专业服务,业务遍布20多个国家,并与众多知名药企建立了长期的合作关系。




点赞
分享
收藏