

Section.01
什么是自噬
自噬(Autophagy)一词源于希腊语“auto”(自我)和“phagy”(吞噬),是细胞在营养缺乏、能量不足或受到应激时,将受损的细胞器、错误折叠的蛋白质或入侵的病原体包裹进双层膜结构的自噬体(Autophagosome),随后与溶酶体融合形成自噬溶酶体(Autolysosome),将其内容物降解并回收利用的过程[1]。
Section.02
自噬的四大类型
自噬不仅是细胞的“清道夫”,更是维持细胞稳态、延缓衰老、抵抗感染和适应环境压力的关键生存机制。自噬并非单一过程,根据底物运送方式和选择性,可分为以下几种类型[2]:
1.巨自噬(Macroautophagy)
最经典的自噬形式。细胞质成分被隔离膜(Phagophore)包裹,形成双层膜的自噬体,再与溶酶体融合完成降解。这是研究最深入的自噬类型。
2.微自噬(Microautophagy)
溶酶体膜直接内陷或突出,将细胞质成分包裹进入溶酶体腔进行降解,无需形成自噬体。
3.分子伴侣介导的自噬(Chaperone-Mediated Autophagy, CMA)
由分子伴侣Hsc70识别带有KFERQ样基序的蛋白质,将其转运至溶酶体膜上的LAMP-2A受体,直接跨膜进入溶酶体降解。这是一种高度选择性的蛋白质降解途径。
4.选择性自噬(Selective Autophagy)[3]
针对特定底物的精准清除:
线粒体自噬(Mitophagy):清除受损线粒体,维持能量代谢稳态
内质网自噬(Reticulophagy):降解应激或多余的内质网
过氧化物酶体自噬(Pexophagy):调控过氧化物酶体数量
核糖体自噬(Ribophagy):回收核糖体亚基
异源自噬(Xenophagy):清除入侵的细菌、病毒等病原体
Section.03
自噬的核心分子机制
自噬是一个高度有序的多步骤过程,涉及30多种自噬相关蛋白(ATG蛋白)的精密协作[3-5]:
第一步:起始(Initiation)—— ULK1复合物的激活
ULK1复合物(由ULK1/2、ATG13、FIP200和ATG101组成)是自噬的"启动开关"。
营养充足时:mTORC1磷酸化ULK1和ATG13,抑制自噬启动
饥饿或应激时:mTORC1活性被抑制,ULK1去磷酸化并被激活,进而磷酸化ATG13和FIP200,启动自噬程序
此外,AMPK在能量不足时可直接磷酸化ULK1的多个位点(如Ser467、Thr574),激活其激酶活性,从而诱导自噬。
第二步:成核(Nucleation)—— PI3KC3复合物组装
ULK1复合物激活后,招募III型磷脂酰肌醇3-激酶复合物(PI3KC3),该复合物核心成员包括VPS34、Beclin1、ATG14L等。
VPS34产生磷脂酰肌醇-3-磷酸(PI3P),招募含有FYVE结构域的蛋白(如DFCP1)和WIPI蛋白,在粗面内质网或线粒体接触位点组装前自噬体结构(PAS),为自噬体形成提供平台。
值得注意的是,AMPK不仅能通过ULK1间接激活PI3KC3,还可直接磷酸化Beclin1(Ser91/94位点)和VPS34,增强复合物的促自噬功能。
第三步:延伸(Elongation)—— 泛素样蛋白连接系统
隔离膜的延伸依赖两个关键的泛素样连接系统:
系统一:ATG12-ATG5-ATG16L1
ATG12在E1样酶ATG7和E2样酶ATG10作用下,与ATG5共价结合
ATG12-ATG5复合物再与ATG16L1结合,定位于隔离膜外表面,促进膜延伸
系统二:LC3脂化(LC3 Lipidation)
新合成的LC3(即LC3-I)在ATG4剪切后,经ATG7(E1)和ATG3(E2)催化,与磷脂酰乙醇胺(PE)共价结合,形成LC3-II
LC3-II嵌入自噬体膜内外两侧,既是自噬体的标志性分子,也参与膜延伸和底物识别
第四步:成熟与降解(Maturation & Degradation)
自噬体通过微管运输至溶酶体,借助SNARE蛋白和Rab GTP酶等介导膜融合,形成自噬溶酶体。溶酶体内的酸性水解酶(如组织蛋白酶)降解内容物,释放氨基酸、脂肪酸等小分子供细胞重新利用。
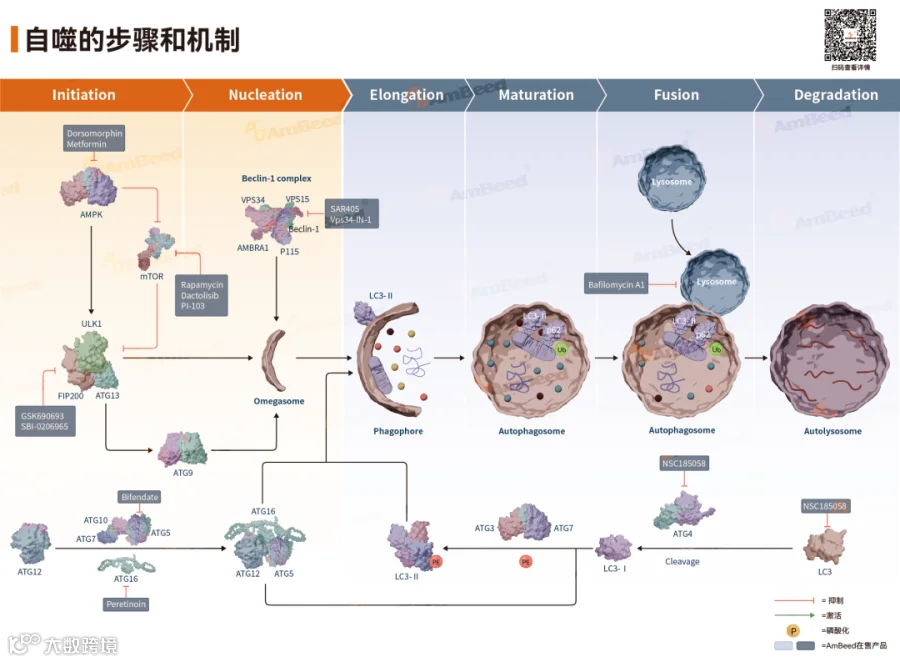
自噬的步骤和机制[4]
Section.04
2026年自噬研究三大前沿突破
突破一:内质网自噬的“信号开关”被破解
内质网是细胞内主要的Ca²⁺储存库,当内质网出现局部损伤时,细胞会启动“内质网自噬”进行清理。但此前,这一过程如何启动、自噬小体的膜从何而来,一直是未解之谜。
2026年4月,中国科学院生物物理研究所张宏团队在《Molecular Cell》发表重磅研究,首次揭示机械感知通道介导Ca²⁺瞬变触发内质网自噬的分子机制,利用超分辨多模态活细胞成像等技术发现[6]:
在长期饥饿、胆固醇稳态失常或高Ca²⁺损伤等应激条件下,内质网中Ca²⁺含量较高的片状亚结构域会扩张并被自噬小体包裹降解
自噬小体膜直接源于内质网重塑,而非传统认为的细胞外膜来源
机械感知通道蛋白PIEZO1和TRPV1富集在高Ca²⁺内质网上,感知钙离子变化并触发局部Ca²⁺释放,形成短暂钙瞬变
该信号触发自噬起始FIP200复合物发生液-液相分离,最终启动内质网自噬
这一发现为神经疾病和癌症等内质网Ca²⁺稳态失衡相关疾病提供了全新干预靶点。
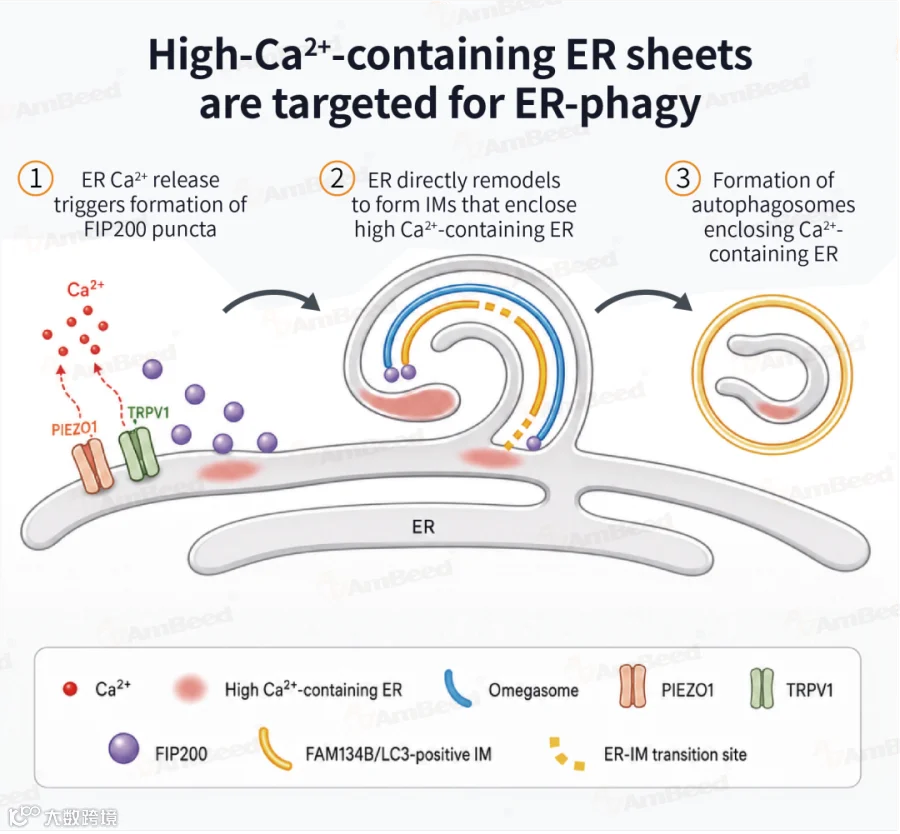
内质网自噬[6]
突破二:自噬体-溶酶体融合的“分子快递”机制阐明
自噬体与溶酶体的融合是决定自噬功能是否完成的“最后一公里”。SNARE蛋白STX17是介导该融合的核心因子,但它在静息状态下广泛分布于内质网、线粒体及胞质,自噬激活时如何精准转运到自噬体膜上,长期困扰学界。
2026年4月30日,北京大学陈建国课题组在《Nature Communications》发表研究,破解了自噬晚期关键步骤的调控密码[7]。研究团队通过CRISPR敲入、单分子追踪等技术首次证实:
向自噬体转运的功能性STX17主要来自胞质库,而非细胞器膜库
饥饿诱导时,STX17第254位赖氨酸(K254)发生乙酰化修饰,由乙酰转移酶GCN5催化,去乙酰化酶SIRT1逆转
K254乙酰化增强STX17与肌动蛋白马达蛋白MYO6的相互作用,驱动胞质STX17向自噬体转运
这一“乙酰化-去乙酰化动态开关”机制,完善了自噬体-溶酶体融合的上游调控通路,为神经退行性疾病和肿瘤治疗提供了新的分子基础。
突破三:代谢-自噬轴成为抗病毒新靶点
2026年4月,南京农业大学周斌团队在《Autophagy》发表研究,揭示病毒如何“劫持”细胞代谢检查点操控自噬[8] 。
二氢乳清酸脱氢酶(DHODH)作为嘧啶合成关键酶,通过调控线粒体自噬影响瘟病毒(Pestiviruses)复制
猪瘟病毒NS4A蛋白将DHODH招募至线粒体,通过其LC3相互作用区(LIR)激活线粒体自噬
该机制具有病毒属特异性:依赖自噬的瘟病毒需要DHODH维持自噬流,而抑制自噬的黄病毒则利用自噬流阻断促进复制外源性嘧啶前体可恢复自噬体-溶酶体融合,为抗病毒药物研发提供了新思路
这一研究将代谢调控与选择性自噬有机结合,开辟了“代谢-自噬轴”抗病毒治疗的新方向。
Section.05
自噬研究的临床转化前沿
2026年,自噬靶向治疗正从"简单激活/抑制"迈向"精准节点调控":
抗衰老药物SRN-901:中美合作研究显示,该药物通过上调线粒体自噬和细胞自噬,使小鼠中位剩余寿命延长三分之一,衰老速度减缓70%[9]
ULK1/2抑制剂ULK-101:在KRAS驱动肺癌中展现比氯喹更强的自噬抑制效果
TFEB激活剂KHS-101:通过mTOR非依赖机制激活自噬,具备血脑屏障穿透性,为神经退行性疾病带来希望[10]
仿生纳米药物MDM:通过磁热效应诱导自噬爆发,增强胶质母细胞瘤铁死亡治疗效果[11]
Section.06
结语
从Ca²⁺信号触发内质网自噬,到乙酰化修饰调控自噬体融合,再到代谢-自噬轴的抗病毒应用,2026年的自噬研究正在回答一个核心问题:如何让细胞的"自我清理"更精准、更高效?
这些突破不仅深化了我们对生命基本过程的理解,更为衰老、癌症、神经退行性疾病和病毒感染等重大疾病的治疗提供了全新策略。自噬,这个细胞最朴素的"生存智慧",正成为现代医学最锋利的手术刀之一。
📚 参考文献
1.Curr Opin Cell Biol. 2025;97:102595.
2.Nat Rev Mol Cell Biol. 2023;24(3):186-203.
3.Mol Cancer. 2024;23(1):22.
4.Cell Death Discov. 2023;9(1):115.
5.Biomolecules. 2024;14(12):1517.
6.Mol Cell. 2026;86(7):1377-1396.e6.
7.Nat Commun. Published online April 30, 2026.
8.Autophagy. Published online April 20, 2026.
9.Drug Des Devel Ther. 2026;20:594895.
10.Int J Mol Sci. 2026;27(2):905.
11.Theranostics. 2026;16(3):1466-1481.
| AmBeed产品推荐 |
|
|
|
|






往期推荐
不止于经典:解密cAMP信号,洞悉其在癌症中的多重调控与治疗新靶点
宫颈癌的“幕后黑手”:HPV是如何一步步“策反”我们身体的?