
第一阶段:单细胞悬液制备与建库(湿实验关键点)
这是单细胞测序最困难、也是最容易引入批次效应的步骤。
1. 组织解离:
将实体组织(如肿瘤、脑组织)消化成单细胞悬液。
难点:必须保证细胞活性高,且不能有太多碎片,否则会影响后续凝胶微滴(GEMs,即凝胶微滴)生成。
2. 细胞捕获与条形码添加:
10x Genomics微流控技术:将单个细胞与含独特条形码(Barcode,即细胞条形码)的凝胶珠包裹在油滴中(GEMs)。
关键原理:每个凝胶珠上有大量引物,这些引物包含:
Cell Barcode:同一细胞的所有mRNA被打上相同的细胞身份证(区分细胞)。
UMI(Unique Molecular Identifier,即独特分子标识符):每条mRNA分子被打上独特的分子身份证(用于去除PCR扩增带来的重复,实现绝对定量)。
3. 反转录与文库构建:
在GEMs内,mRNA被反转录为cDNA,此时cDNA已经带上了细胞条形码和UMI。
破油后,进行PCR扩增,然后构建测序文库。
第二阶段:测序与数据初步处理
测序完成后,你拿到的依然是FASTQ文件,但解读方式完全不同。
1. 原始数据质控
内容:与普通测序类似,检查测序质量、GC含量等。
软件:FastQC。
2. 数据初处理(Cell Ranger 核心步骤)
这是单细胞分析特有的第一步,通常使用 10x Genomics 官方的 Cell Ranger count 管道。
输入:FASTQ 文件。
参考基因组:需要提供基因组的FASTA文件和GTF注释文件。
主要任务(解复用):
比对:将读段比对到参考基因组。
细胞区分:根据条形码区分哪些GEMs里真的有细胞,哪些只是空的油滴(背景噪声)。
UMI计数去重:利用UMI去除PCR重复,统计每个基因在每个细胞中实际有多少个独特的mRNA分子。
产出文件(关键):
表达矩阵(Filtered Feature-Barcode Matrix):一个巨大的表格,行是基因,列是细胞,中间的数字是UMI计数。这是后续所有分析的起点。
BAM文件:比对文件,可用于可视化验证。
Web Summary HTML:包含细胞数、测序饱和度、比对率等关键质控指标。
第三阶段:高级分析(数据挖掘)
拿到表达矩阵后,接下来的分析通常使用 R语言 + Seurat包 或 Python + Scanpy包 进行。这是单细胞分析最有趣的部分。
1. 数据质控与过滤
并不是所有检测到的“细胞”都是好细胞。
过滤标准:
线粒体基因比例过高(通常 > 10-20%):表明细胞质膜破裂,细胞正在死亡或已死亡。
检测到的基因数过少:可能是空液滴或死细胞。
检测到的基因数过多:可能是一个液滴包裹了两个细胞(双细胞或多细胞)。
软件:Seurat / Scanpy。
2. 数据标准化与归一化
目的:消除测序深度(即每个细胞测到的总UMI数不同)带来的技术误差。
方法:将每个细胞的UMI计数除以该细胞的总UMI数,再乘以缩放因子(如10,000),最后进行对数转换(LogTransform)。
3. 特征选择与降维
高变基因筛选:找出在细胞间表达差异最大的基因(这些基因定义了细胞的身份)。
主成分分析:将数千个基因的信息压缩成几十个主成分,去除噪音。
非线性降维:
t-SNE / UMAP:将高维数据投射到二维平面,以便可视化。
4. 细胞聚类
目的:让计算机根据基因表达谱的相似性,自动将细胞分群。
结果:每个细胞被分配一个“簇”ID(如Cluster 0, Cluster 1...),通常认为每个簇代表一种细胞类型或状态。
5. 差异表达分析与细胞类型注释
找标记基因:找出每个簇(Cluster)相对于其他簇高表达的基因(如Cluster 0高表达CD3D,Cluster 1高表达CD79A)。
人工注释:根据标记基因,结合生物学知识,给每个簇命名(如 Cluster 0 = T细胞,Cluster 1 = B细胞)。
辅助工具:SingleR(自动注释)、CellMarker数据库。
6. 下游高级分析(根据研究目的定制)
轨迹推断/拟时序分析:
目的:研究细胞动态变化过程,如T细胞分化、肿瘤演进。
软件:Monocle 3、Slingshot。
原理:既然无法追踪同一个细胞在不同时间点的变化,就假设细胞处于不同的成熟阶段,将它们按基因表达的相似性排列成一条“轨迹”。
细胞通讯分析:
目的:研究不同细胞类型之间如何通过配体-受体对进行交流。
转录因子调控网络:
目的:找出驱动细胞状态转变的核心转录因子。
差异丰度分析:
目的:比较疾病组与对照组中,某种细胞类型的比例是否发生显著变化。
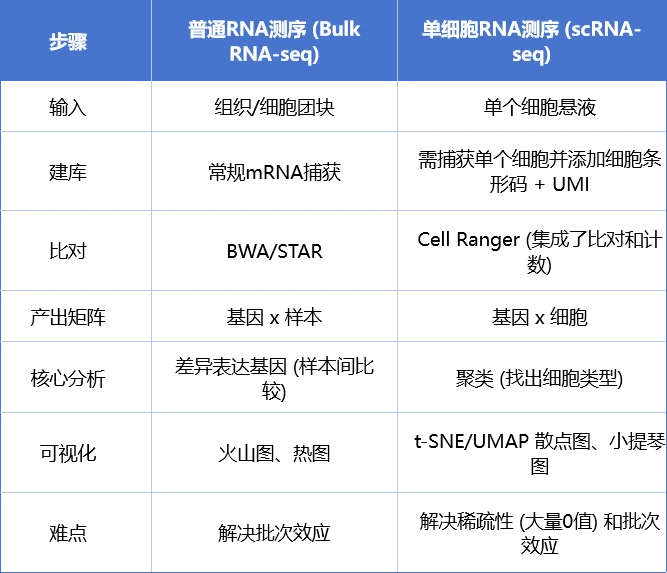
单细胞 vs 普通转录组流程对比
需要留意的关键点
稀疏性:单细胞数据非常稀疏,通常有90%以上的基因表达量为0,这是正常的(因为每个细胞内的mRNA本来就很少)。
批次效应:不同时间、不同人做的实验,技术误差可能比生物学差异还大,需要用 Harmony 或 Seurat IntegrateData 等方法进行校正。
计算资源:处理几万个细胞的数据,通常需要较大的内存(32GB以上可能不够),必要时需要使用云服务器。
目标
应用科学技术,让人民的生活更美好。
联系我们,打造非凡的人工智能 & 多肽产品
muyonglin@aiptide.com
”
关注我们
ATTENTION US

<了解更多信息>
- 作品说明 -
素材 | 智普肽德
文案 | 智普肽德
图片 | 智普肽德