尊敬的读者们大家好,在合集第二、三篇内容中,已详细介绍了“钠离子电池—阴极”的相关专业知识。本篇将为大家普及“钠离子电池—阳极”的相关专业知识。
在前一部分的详细讨论之后,我们现在转向正极的对应部分:阳极,即电池的负极端。要开发有前景的负极材料,它必须满足以下标准。首先,材料中的某一元素需要具备较低的原子质量和低密度,同时能够容纳大量钠离子/单位化学式,这些特性可以确保稳定的高质量比容量(mAh·g⁻¹)和体积比容量(mAh·cm⁻³)。其次,该材料不应具有高反应性,且不易溶解于电解质溶剂中。第三,材料的电势应接近钠金属,且不随钠含量变化,以保证与正极结合时的工作电压稳定。第四,材料必须环保、危害性小,并具备高的电子和离子导电性。除了这些基本要求,还存在其他实际挑战,例如在高截止电压下运行可能导致材料体积膨胀和粉碎,能量密度低,以及初始循环中库伦效率低等问题。此外,电动汽车市场对SIB负极能量密度的要求不断提高,以期匹配锂离子电池的优势,这带来了额外的挑战。
与LIB不同,其中Li金属可以作为负极材料,Na金属不能用作SIB的负极,因为它会导致严重的树枝晶形成,对电解质具有高反应性,并且与Li(180.5 °C)相比,它具有低熔点(97.7 °C),这进一步引发了安全问题。尽管正在探索许多其他材料作为SIB的负极材料,但根据报道的反应机制(图17),它们可以大致分为1)转化材料,2)嵌入/嵌入型,和3)合金型。另一类材料属于碳基和基于氧化钛的材料;然而,它们可以处理为嵌入型负极材料。转化型负极材料可以被定义为主要由过渡金属(TM)及其衍生物(例如,氧化物、硫化物和硒化物)组成的材料,它们能够储存大量的钠离子的方式,主要是通过转化反应形成Na富集的金属化合物,如Na3P、Na15Sn4和Na3Sb。在第一份报告中,Poizot观察到,纳米尺寸的TM-O可以通过完全不同于Li嵌入/脱出的反应机制储存大量的Li离子:MxOy + 2yLi+ + 2ye− ↔ xM0 + yLi2O。嵌入/嵌入化合物可以被定义为主体材料接受客体物种进入其晶体结构的能力,而不会过多地干扰晶体参数,如单位体积、晶体相或平面之间的距离。客体(嵌入)物种的数量由电极/电解质界面达到的热力学平衡控制。到目前为止,嵌入型化合物是最受欢迎的SIB材料,甚至超越了锂离子电池技术。嵌入化合物内部的结构相稳定性和有序性显著影响电池性能,包括决定电压曲线、扩散动力学和衰退机制。对于合金型材料,可以简单地理解为一些来自XIV和XV组的金属和非金属元素(如Si、Ge、Sn、Pb、P、As、Sb和Bi)通过合金化或脱合金化反应,与钠形成Na-M二元金属化合物。这种合金反应被认为可以提供优秀的理论容量,但由于材料的体积膨胀巨大,导致电极材料自粉碎,从而引发严重的容量和电压衰减及循环性差的问题。更严重的是,为了利用已经建立的锂离子电池市场,SIB必须克服Na+(1.02 Å)较大离子半径导致的慢扩散动力学,这限制了快速Na+储存。因此,必须投入更多的努力来提高SIB的电化学性能。以下部分将关注SIB负极材料的最新进展和现有挑战。
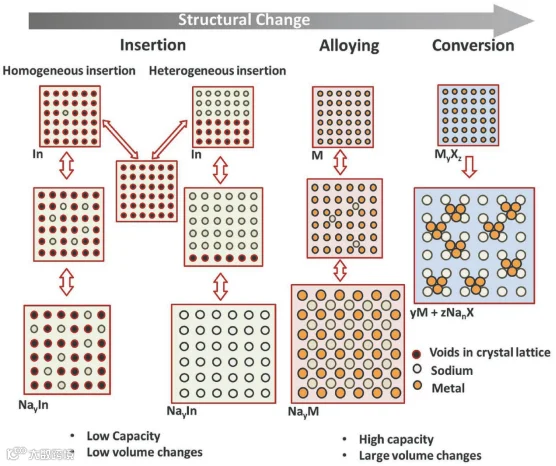
图17. 已报道的Na+在各类阳极中储存的机理示意图。
4.1 转化型材料
一些过渡金属(TM),以及它们的氧化物(TMOs)、硫化物(TMSs)和磷化物(TMP),可以通过转化反应机制储存Na+。与嵌入型和合金型不同,其中碱金属离子在主体晶格中可逆地进出,转化反应的特别之处在于它们涉及将一个或多个原子种类转化为主体晶格,形成新的化合物。由于它们的高理论容量,基于转化的材料是SIB的潜在候选材料;然而,它们的主要电化学反应发生在低电压下,导致这些材料主要作为SIB的负极应用。此外,这些转化材料也容易发生严重的体积膨胀,可能在钠化/脱钠过程中接近(200-300)%而损坏电极,这通常会导致导电网络失去接触,随后导致严重的容量衰减。此外,Na+更大的离子半径与其缓慢扩散动力学对储存Na+构成了最大的威胁。随着SIB研究的进展,研究人员已经展示了碳导电技术可以显著提高这些基于转化的负极的导电性。本节重点介绍了SIB转化型材料的重大突破和现有挑战。
4.1.1 过渡金属氧化物
转化材料的概念最初由Alcantara等人在尖晶石NiCo2O4氧化物中引入作为SIBs的负极材料。作者提出,钠和金属氧化物之间的可逆转化反应可以通过以下反应描述:NiCo2O4 + 8Na → Ni + 2Co + 4Na2O。从那时起,各种研究小组探索了一系列其他的TMOs。一些值得注意的氧化物是铁的氧化物Fe3O4和Fe2O3;在这个系列中,钴的氧化物,如CO3O4,锡氧化物SnO和SnO2,镍氧化物如NiO和NiO-Ni,以及锰氧化物Mn3O4占据了重要的位置。
铁氧化物:早在2010年,Komaba等人报道了Fe3O4可以作为SIB的负极材料,遵循在电压范围(1.2-4.0)V内的嵌入反应机制。然而,Fe3O4具有作为嵌入和转化材料的独特特性,使其成为TMO类别中最被探索的材料。在转化反应过程中,初始循环期间提供了≈643/366 mAh·g-1的放电/充电比容量,在10个循环后保持率为65%,并且显示了57%的良好库伦效率。随着时间的推移,提出了几种改进策略来提高这种材料的性能,如添加沥青碳作为涂层来包裹纳米尺寸的Fe3O4颗粒,并将C/Fe3O4的纳米复合物与CNT锚定,提供了321/440 mAh·g-1的首次充放电容量,库仑效率为73%。最近,石墨烯气凝胶包裹的Fe3O4,表示为Fe3O4/@rGA,被合成用作SIB的高性能柔性负极材料。这种合成的负极在电流密度为100 mA·g-1时提供了220 mAh·g-1的初始容量,在电流密度为10 A·g-1时提供了82 mAh·g-1的良好可逆容量。在水热合成过程中产生的大量多孔结构提高了电导率,而石墨烯的包裹避免了颗粒的聚集,并防止了Fe3O4颗粒和电解质在连续循环中的直接接触。此外,石墨烯气凝胶的包裹也缓解了体积膨胀,并抑制粉碎。调整Fe3O4颗粒的形态是改进这种负极材料性能的另一个工具。Ren和他的同事们合成了一个棱柱形的核壳修饰在N掺杂的碳上Fe3O4,并散布在石墨烯上,表示为Fe3O4@NCm/还原氧化石墨烯(rGO)。这种合成方法被认为可以改善Fe3O4的低电导率、体积膨胀和颗粒聚集。由于N掺杂的碳,这种负极在电流密度为1000 mA·g-1的2500个循环中提供了237 mAh·g-1的容量,在全电池配置中,容量为192 mAh·g-1。有几个因素导致了这种性能的提高。首先,碳中的N掺杂带来了更多的活性位点,显著提高了电导性。其次,空心结构暴露出更多的活性位点参与电化学反应。第三,碳保护和它与石墨烯的协同作用进一步赋予了这种复合负极稳定性,大大缓解了颗粒粉碎。
锡氧化物:由于其高理论容量、低成本、丰富、环境友好、适当的工作电位和多功能应用,锡氧化物(SnO2)及其化合物是有前途的材料。SnO2是一种可以同样应用于LIB和SIB负极的材料。然而,由于在嵌入过程中的严重体积膨胀,导致不可逆的容量损失并缩短循环寿命,其实际应用受到阻碍。最近,研究了SnO2及其衍生物在电池负极材料中的应用。例如,Gu等人通过原位透射电子显微镜(TEM)探测了SnO2纳米线(NWs)的失效机制。Na在SnO2中的初次嵌入伴随着位移反应,导致在Na2O基体内形成无定形的NaxSn NPs。在进一步钠化过程中,NaxSn晶化为Na15Sn4(x = 3.75),而在脱钠过程中,NaxSn转化为Sn NPs。在钠化过程中,体积膨胀巨大,纳米线从直径67 nm膨胀到145 nm。在Na脱出过程中,发生相反的情况,NWs收缩(≈109 nm),并形成几个孔洞,这些孔洞显著限制了电导率,从而导致稳定性较差。后来,Wang等人通过水热法在碳布集流体上直接生长出交错的SnO2纳米片结构,通过电沉积涂上聚吡咯(PPy),表示为SnO2@PPy,这种负极提供了1172.1 mAh·g-1的初始放电容量,在0.1 A·g-1的300个循环后,容量保持率仍为85%。人们认为这种涂层能够解决体积膨胀问题,并在更大程度上提高电导率。SnO2@PPy的测量Rct(225.6 Ω)远低于SnO2(482.7 Ω),表明PPy涂层带来了更好的导电性。然而,实现SnO2的理论容量(1558 mAh·g-1)仍有很长的路要走,必须进行研究努力才能实现。最近,采用了一种独特的设计策略,直接在原位形成的N掺杂的分支TiO2/C纳米纤维(NBT/C@SnO2 NFs)上生长SnO2 NSs。这种无粘结剂的负极在200 mA·g-1的500个循环后提供了高可逆容量(420.7 mAh·g-1),并在非常高的电流密度(2000 mA·g-1)下仍显示出330.6 mAh·g-1的容量。电化学性能的改善归因于在低温合成过程中获得的类似无定形的结构,这种结构创造了一个无序和松散的结构,以适应Na+嵌入过程中的体积膨胀。此外,在随机取向的碳层和层状石墨烯结构之间形成的微/纳米孔将加速离子扩散和电子传输,从而增加了电导率。而在碳中掺杂N会产生一些缺陷位点,这些位点提供了更多的扩散通道和更多的Na+存储活性位点。为了进一步提高SnO2的电化学性能,最近,通过固态合成法合成了锡氧化物锚定在S和N共掺杂的碳中(SnO2@SNC)。这种独特设计的结构在1000个循环结束时以2 A·g-1提供了600 mAh·g-1的可逆放电容量。这种复合材料的出色性能归因于电极的增强电导率,以及由碳材料确保了反应可逆性,同时碳的存在进一步抑制了由转化过程引起的容量衰减。
钴氧化物:基于钴的材料由于其高催化能力、非贵金属特性和经济实惠的特点,显示出潜在的能源应用可能性,特别是因为其高理论容量(890 mAh·g⁻¹)在电池中的应用受到关注。尽管这些材料具有经济效益和高理论容量,但它们受限于有限的循环寿命和在钠化/脱钠过程中的大容量膨胀。为减少这些问题,如粒径减小和将它们约束在碳质基体中,已经进行了多项努力,以推动其商业化。首次提出一种应用于CO3O4与Na+的可逆转化反,通过XRD和CV循环测得为CO3O4 + 8Na+ + 8e- ↔ 4Na2O + Co。基于离位XRD分析,观察到转换反应在首次放电到0.01 V时不完全,它在进一步的循环中继续。作者还推测,在循环过程中,放电容量将一直增加直到转换反应完成,然后容量可能会保持或下降。在转换反应之后,这种材料展示了一个可逆放电容量为447 mAh·g-1,是其在第50个循环后保持率为初始的86%。后来,Rahman等人进一步修改了这种材料,并通过结合熔盐法和液体等离子技术,合成了Co3O4/CNTs。这种复合结构具有几个优点,1)应力/应变大大减轻,2)它可以容纳大的体积变化并抑制其带来的影响,3)CNT基体提供电子导电性和稳定性,以及4)减小离子或电子传输的时间。这种复合结构在100个循环后以50 mA·g-1的电流密度下提供了403 mAh·g-1的可逆容量,并且在1.6/3.2 A·g-1处显示出212/190 mAh·g-1的倍率能力。因此,这种复合结构作为负极材料提供了巨大的潜力。后来,报道称一种由Co3O4 NSs和Co3O4/碳同素异形体(Co3O4/C)组成的复合物可以作为负极材料。这种复合结构(Co3O4/GNSs)在电流密度为100 mA·g-1的100次循环后提供了436.9 mAh·g-1的容量。添加石墨烯NSs(GNSs)带来高灵活性和导电性,可以缓冲Co3O4在重复循环中的体积变化,并通过降低Rct提升较好的电导率。
锑氧化物:在众多氧化物材料中,氧化锑也是用作钠离子电池(SIB)负极的有前景的候选材料。由于其高理论容量和电化学活性,氧化锑在能源应用中受到了极大的关注。然而,它们也受到了巨大体积膨胀和较差电导率的困扰,在最近的一项研究中,合成了嵌入氮掺杂碳纤维中的SbO2/Sb2O3纳米粒子用作负极材料。这种复合物(SbO2/Sb2O3@NC)在电流密度为100 mA·g-1时提供了307 mAh·g-1的可逆容量。这种优越的电化学性能归因于碳纤维限制了大的体积变化,以确保耐久性。后来,Liu等人合成了掺硼的Sb/SbO2@rGO复合物,这种复合物在电流密度为50 mA·g-1、经过100个循环后,显示出346 mAh·g-1的可逆容量,保持率为106%。这种改进的电化学特性是通过硼掺杂引入的,这显著地扩大了SbO2的晶格间距,从而加速了Na+运输的扩散。最近,Mxenes也已经与Sb2O3结合进行测试,以合成用于储钠的Sb2O3/Ti3C2Tx复合物。这种复合结构展示了增强的结构稳定性,在2 A·g-1的倍率容量为295 mAh·g-1,以及在100 mA·g-1经过100个循环后的放电容量为472 mAh·g-1。优越的电化学性能,特别是倍率性能,归因于Sb2O3和Mxene鳞片之间的协同作用。具有高导电性的Mxene Ti3C2Tx鳞片与提供给Sb2O3纳米粒子高导电性的3D结构链接。这种特性改善了充电转移阻抗,并反映在倍率性能中。另一方面,Sb2O3的亚纳米粒子显著抑制了Ti3C2Tx鳞片的重叠,这是在能源存储应用中使用MXene时常见的问题。此外,在自组装过程中产生的空隙可以容纳Sb2O3钠化过程中的体积膨胀,从而在几个循环中保持结构稳定性。尽管Mxene提供了几个好处,但必须进行理论研究,以理解多个脱钠状态期间的扩散动力学和形成能(f.u.),以确定稳定性,并进一步改善在SIB中使用时的电化学性能。
氧化铜:在寻找氧化物材料作为负极应用的竞赛中,铜氧化物因其丰富的地球分布、高理论容量和更好的化学稳定性而备受关注。然而,类似于其他过渡金属氧化物,铜氧化物的电导率较差且体积膨胀明显,这严重损害了电极的完整性,导致稳定性和倍率性能不佳。为应对这些缺点,将负极材料进行纳米结构设计以缩短电子和离子的传输路径,是相较于块体材料的一项关键技术。例如,通过水热处理得到的带CMC粘结剂的CuO纳米片已被证明是钠离子电池的理想负极。这个负极在电流密度为100 mA·g-1时显示出562.2 mAh·g-1的初始放电容量,保持率为70.1%。作者还研究了使用其他粘结剂(如PVDF)的电化学性能,在相似条件下能够提供初始容量为380.9 mAh·g-1。这种电化学性能的差异是由于PVDF粘结剂的电极产生了几道裂纹,这加速了粒子的解体和结构的破败,而在CMC中则没有看到这样的裂纹。这个发现也解释了并暗示粘结剂在决定钠离子电池的电化学性能中可能有多重要。如前一部分所解释的,在转化材料中,转化反应通常在第一周期中没有完成,特别是在核壳负极中,这往往导致"未反应的核",导致严重的容量衰减。这里必须提到的是,大多数负极材料经历了从结晶性到低结晶性,甚至到无定形态的相变,因此X射线衍射在检测循环过程中的结构演变时失效。在这种情况下,X射线吸收光谱或原子对分布函数可以作为一个有效的工具来检测阶段的变化,甚至是在无定形状态下。为了正确理解嵌入反应机制,半电池组分是一个有效的工具。CuO/Na的初始钠化提供了一个较差的电化学性质(充放电:132/330 mAh·g-1),这远低于Li对应物(CuO/Li:665 mAh·g-1)。这种较差表现的一个已知原因是由于钠离子的较大离子半径导致的较慢的嵌入动力学。然而,它们的循环伏安曲线显示了一种非常规的模式,在钠离子的低电压区域会发生大的充放电,而在Li离子电池中则不会。三个还原峰位(2.21,1.09和0.32)V分别对应于中间铜氧化物相(Cu2O),接下来是Cu2O转化为Cu,和固体电解质界面膜的形成,而氧化峰位(1.26和2.12)V分别对应于Cu2O和Cu的生成。与此同时,在Li离子电池中,主要的还原峰位在1.03/0.63 V,分别对应于中间的Cu2O阶段/Cu,而氧化峰位在0.80/2.46 V,对应于SIB中类似的阶段(图18b)。SIB中出现的非常规平坦放电和大的极化引发了一些需要立即关注的问题。除了已知的事实,即所有负极材料在Na+的嵌入/脱出过程中都会膨胀/收缩,负极粒子在放电的最后阶段仍然“未反应”,在0.1 V以下没有微观结构变化。这种出乎意料的行为与锂离子电池相反,后者在锂化过程中,特别是在0.1 V以下,会出现裂纹和断裂。采用X射线纳米断层成像展示了电池颗粒的特写视图(图18c,d)。这些图像显示了一个完美的核壳结构,除了顶部和底部的部分,在那里组分完全钠化为单一成分。根据这些发现,作者提出了一个科学概念,即微观结构的完整性可能在钠化过程中导致未反应核心的形成,这进一步限制了Na+的嵌入,导致循环效率低下。
除了氧化物,硫化物是另一种广泛用于储能的基本材料。这些材料引起了极大的关注,因为它们通过转化反应机制表现出高理论容量,在存在过渡金属氧化物(TMO)的情况下,它们的储存容量可以进一步提高。在化学上,过渡金属-硫(TM-S)键稍微弱于过渡金属-氧(TM-O)键,这实际上更有利于它们与钠离子进行转化反应,公式如下:TMSx + 2xNa ↔ TM + xNa2S。这种TMS的转化路径赋予了它们更好的机械稳定性,因为体积变化较小,由于在(脱)钠化过程中Na2S对Na2O的可逆性增强,库仑效率也得到改善。因此,已有多种TMS复合物被探索作为钠离子电池的负极材料。
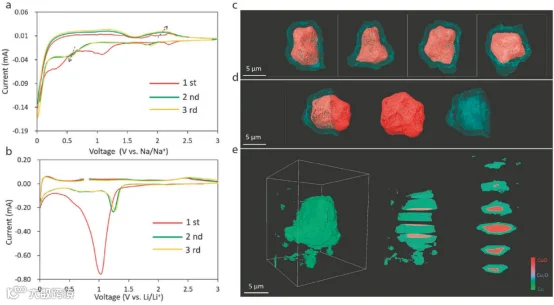
图18.a) SIBs的循环伏安图。b) LIBs的循环伏安图。X射线纳米断层扫描显示SIBs中不反应的CuO粒子核的现象。c) 不同角度的三维成像。d) 不反应核的切割截面。e) SIBs中不反应的CuO核的切片视图。
铁硫化物:铁硫化物(FeS)是一种受欢迎的成员,由于其低毒性和环境丰富性,以及更重要的是其低成本优势而被广泛用作负极材料。从理论上讲,FeS可以提供606 mAh·g-1的容量,并属于P63/mmc (a = b = 3.43 Å,c = 5.79 Å)空间群系列。半电池中的转化反应可描述为:2FeS + xNa + xe- ↔ Fe + NaxFeS2 (x ≤ 2) 和 NaxFeS2 + (4-x)Na+ + (4-x)e- ↔ Fe + 2Na2S。姚等人合成了具有不同多巴胺涂层含量的花状FeS/C复合物。其中,FeS/C-2电极表现出最优性能,分别在1/4 A·g-1时提供809.7/403.2 mAh·g-1的可逆容量, 100个循环周期保持77.3%的容量和高倍率性能(10C时为368.3 mAh·g-1)。电化学性能的提高归功于碳涂层提高了电导率(电子/离子路径的缩短)并且还可以容纳体积膨胀。如前所述,氮掺杂是另一种改善性能的策略,刘等人通过使用Fe(NO3)3和(NH2)2CO混合物的原位化学转化途径合成了N掺杂的碳复合物(FeS/NC)。这种合成技术具有一个独特的功能,允许通过原位生长的N掺杂碳来包覆单个FeS纳米颗粒,然后嵌入超薄碳片内,在200/1000 mAh·g-1电流下分别提供511/326 mAh·g-1的容量,可循环100次以上。性能的提升归因于原位碳涂层充当海绵以容纳体积膨胀,而相互连接的超薄碳片增加了电导率。核壳结构也可选用以改善电化学性能,如最近由徐小组合成的3D石墨烯封装核壳FeS@C纳米复合物。这种合成方法(3DG/FeS@C)在80个周期后提供了632 mAh·g-1的容量,以及1/6 A·g-1下分别的363.3/152.5 mAh·g-1的良好倍率性能。作者声称,即使在1 A·g-1电流下循环300次后,这个负极仍保持了97.9%的容量,这是迄今为止基于FeS化合物的最佳报告。这种多孔的3D核-壳结构提供了一个优秀的导电网络,显著提高了电导率,显示出一个小的Rct(≈250 Ω),而浸渍的3D石墨烯网络为离子/电子提供了一个缩短的扩散路径,坚固的封装(碳和石墨烯层)缓解了体积膨胀/收缩,防止了FeS颗粒的聚集。总的来说,这些特性改善了电化学性能。
钼硫化物:二硫化钼 (MoS2) 是一种二维层状、廉价且地壳富集的材料,它在能量转换反应中有较广泛的应用,如水分解反应。MoS2通常是半导体,因此它的电子导电性较差,倍率能力也稍低。结构上它由两层硫原子之间的一个钼原子层组成,六个硫原子与钼配位形成一个MoS6八面体。这些层之间有微弱范德华力,因此可以像石墨那样接纳阳离子。理论上,MoS2可以提供670 mAh·g-1的容量。更具体地说,理论计算显示,当Na的范围在(0-1)时,Na+在MoS2中的嵌入在热力学上对Na的相分离有利。在对MoS2和MoO2进行比较研究时,发现由于MoS2的层间距离比MoO2大,所以MoS2先与Na+进行嵌入反应,然后进行转化反应。此外,MoS2的键稍弱于MoO2,这推动了前者进行转化反应。理论计算还预测,两种材料MoS2/MoO2的转化反应吉布斯自由能分别为-483.3/-220.0 kJ·mol-1,这支持前者与Na反应更容易。转化反应由金属阳离子和阴离子之间的结合能决定;MoO2具有较小的板间距离和强的MO-O键,与弱结合的Mo-S相比,不利于Na+的嵌入。在最近的一篇报告中,通过使用一个1T (三方晶系) 和2H (六方晶系) 阶段合成了双相MoS2 (DP-MoS2)。作者认为,通过结合双相,可以解决MoS2 (导电性差且在循环过程中结构崩溃)的固有问题。合成的材料在0.5 A·g-1的电流下,经过200个循环,提供了300 mAh·g-1的容量,并且具有极好的倍率能力,即在2 A·g-1下约220 mAh·g-1。此外,与Na3V2(PO4)3作为正极的全电池,它在0.5 A·g-1时能提供210 mAh·g-1的容量。DP-MoS2的较高电导率 ((2.23x10-11)-(3.60x10-10)) cm2·S-1 促进了Na+的快速扩散动力学,从而保证了高倍率性能。DFT计算显示,1T和2H相界面处形成的固有电场吸引Na+朝向2H-MoS2,这加快了Na+的迁移速度。最近,设计了MXene-MoS2异质结构,目的是通过缩短扩散路径来显示Na+的更快传输。 这种稳定的异质结构进一步被认为能够带来连续的扩散通道,以实现更快的电子传递。合成的负极示出了0.2 A·g-1电流下出色的可逆容量315 mAh·g-1 和在1000个循环周期后2.0 A·g-1电流下的220.0 mAh·g-1。MXen显著降低了Na原子的吸附能量障碍(0.078 eV),这比MoS2@MoS2(1.28 eV)或MXene@MoS2(0.11 eV)等其他界面要低得多。MXene显著降低了MoS2的带隙以显著提高导电性,这些优秀的电化学性能的提高是由MXene保证的。
钒硫化物:钒硫化物(VS2)的电子传输效率比MoS2高得多,由于它们的金属性质,因此经常被认为是具有重要意义和适合作为负极材料的材料。VS2的理论研究表明,可以达到高达466 mAh·g-1的容量,输出电压低(0.48 V),其扩散系数可以达到与体相MoS2相比的七个数量级。负极的倍率性能可以直接通过Na+的扩散动力学来解释,结果表明,VS2中的Na+扩散动力学比MoS2(170 meV)低得多。此外,钠离子的活化能垒也比MoS2(780 eV)低。这些特性支持了使用VS2作为负极材料比MoS2有更多的优点的观点。然而,可用容量(≈250 mAh·g-1)比理论预测的要低得多,也比碳基材料低得多,这一直是使用它作为SIB电池负极的一个重要挑战。通过溶剂热法合成了具有层状结构的VS2纳米片,其在0.1 A·g-1下的容量为600 mAh·g-1,同时在循环保留性上表现出优异的性能(700个周期),在(2和5)A·g-1下分别为(83%和87%)。它也展示出优秀的倍率能力,在非常高的电流密度(20 A·g-1)下放电容量为277 mAh·g-1。这种卓越的电化学性能是由于赝电容机制,特别是在高电流密度下。花状VS2纳米片有利于电解液的渗透,足够增加的电解液界面使离子传输更快。值得注意的是,VS2纳米片提供了600 mAh·g-1的容量,这表明近2Na+参与到电化学反应中,这比其他参与硫酸亚铁电池的负极材料要多得多。通过氮掺杂,可以进一步改进这种花状VS2结构的电化学性能。例如,通过溶剂热过程在还原石墨烯上生长得到花状VS2/N掺杂碳(VS2/N-C)具有扩大的(001)面,然后进行碳化技术。合成的VS2/N-C@rGO纳米杂化物在1.0 A·g-1下提供了407 mAh·g-1的可逆容量,高倍率容量(8 A·g-1下的273 mAh·g-1),并且具有非常长的稳定性(1400个循环)。扩大的层间距(1.02 nm),显著降低了离子扩散壁垒,花状结构提供了数个活性位点,并且可以容纳在循环过程中的大体积膨胀,这两者共同改善了这个负极的电化学性能。
4.1.2 过渡金属硫化物
除了氧化物,硫化物是另一种广泛用于储能的基本材料。这些材料引起了极大的关注,因为它们通过转化反应机制表现出高理论容量,在存在过渡金属氧化物(TMO)的情况下,它们的储存容量可以进一步提高。在化学上,过渡金属-硫(TM-S)键稍微弱于过渡金属-氧(TM-O)键,这实际上更有利于它们与钠离子进行转化反应,公式如下:TMSx + 2xNa ↔ TM + xNa2S。这种TMS的转化路径赋予了它们更好的机械稳定性,因为体积变化较小,由于在(脱)钠化过程中Na2S对Na2O的可逆性增强,库仑效率也得到改善。因此,已有多种TMS复合物被探索作为钠离子电池的负极材料。
铁硫化物:铁硫化物(FeS)是一种受欢迎的成员,由于其低毒性和环境丰富性,以及更重要的是其低成本优势而被广泛用作负极材料。从理论上讲,FeS可以提供606 mAh·g-1的容量,并属于P63/mmc (a = b = 3.43 Å,c = 5.79 Å)空间群系列。半电池中的转化反应可描述为:2FeS + xNa + xe- ↔ Fe + NaxFeS2 (x ≤ 2) 和 NaxFeS2 + (4-x)Na+ + (4-x)e- ↔ Fe + 2Na2S。姚等人合成了具有不同多巴胺涂层含量的花状FeS/C复合物。其中,FeS/C-2电极表现出最优性能,分别在1/4 A·g-1时提供809.7/403.2 mAh·g-1的可逆容量, 100个循环周期保持77.3%的容量和高倍率性能(10C时为368.3 mAh·g-1)。电化学性能的提高归功于碳涂层提高了电导率(电子/离子路径的缩短)并且还可以容纳体积膨胀。如前所述,氮掺杂是另一种改善性能的策略,Liu等人通过使用Fe(NO3)3和(NH2)2CO混合物的原位化学转化途径合成了N掺杂的碳复合物(FeS/NC)。这种合成技术具有一个独特的功能,允许通过原位生长的N掺杂碳来包覆单个FeS纳米颗粒,然后嵌入超薄碳片内,在200/1000 mAh·g-1电流下分别提供511/326 mAh·g-1的容量,可循环100次以上。性能的提升归因于原位碳涂层充当海绵以容纳体积膨胀,而相互连接的超薄碳片增加了电导率。核壳结构也可选用以改善电化学性能,如最近由徐小组合成的3D石墨烯封装核壳FeS@C纳米复合物。这种合成方法(3DG/FeS@C)在80个周期后提供了632 mAh·g-1的容量,以及1/6 A·g-1下分别的363.3/152.5 mAh·g-1的良好倍率性能。作者声称,即使在1 A·g-1电流下循环300次后,这个负极仍保持了97.9%的容量,这是迄今为止基于FeS化合物的最佳报告。这种多孔的3D核-壳结构提供了一个优秀的导电网络,显著提高了电导率,显示出一个小的Rct(≈250 Ω),而浸渍的3D石墨烯网络为离子/电子提供了一个缩短的扩散路径,坚固的封装(碳和石墨烯层)缓解了体积膨胀/收缩,防止了FeS颗粒的聚集。总的来说,这些特性改善了电化学性能。
钼硫化物:二硫化钼 (MoS2) 是一种二维层状、廉价且地壳富集的材料,它在能量转换反应中有较广泛的应用,如水分解反应。MoS2通常是半导体,因此它的电子导电性较差,倍率能力也稍低。结构上它由两层硫原子之间的一个钼原子层组成,六个硫原子与钼配位形成一个MoS6八面体。这些层之间有微弱范德华力,因此可以像石墨那样接纳阳离子。理论上,MoS2可以提供670 mAh·g-1的容量。更具体地说,理论计算显示,当Na的范围在(0-1)时,Na+在MoS2中的嵌入在热力学上对Na的相分离有利。在对MoS2和MoO2进行比较研究时,发现由于MoS2的层间距离比MoO2大,所以MoS2先与Na+进行嵌入反应,然后进行转化反应。此外,MoS2的键稍弱于MoO2,这推动了前者进行转化反应。理论计算还预测,两种材料MoS2/MoO2的转化反应吉布斯自由能分别为-483.3/-220.0 kJ·mol-1,这支持前者与Na反应更容易。转化反应由金属阳离子和阴离子之间的结合能决定;MoO2具有较小的板间距离和强的MO-O键,与弱结合的Mo-S相比,不利于Na+的嵌入。在最近的一篇报告中,通过使用一个1T (三方晶系) 和2H (六方晶系) 阶段合成了双相MoS2 (DP-MoS2)。作者认为,通过结合双相,可以解决MoS2 (导电性差且在循环过程中结构崩溃)的固有问题。合成的材料在0.5 A·g-1的电流下,经过200个循环,提供了300 mAh·g-1的容量,并且具有极好的倍率能力,即在2 A·g-1下约220 mAh·g-1。此外,与Na3V2(PO4)3作为正极的全电池,它在0.5 A·g-1时能提供210 mAh·g-1的容量。DP-MoS2的较高电导率 ((2.23x10-11)-(3.60x10-10)) cm2·S-1 促进了Na+的快速扩散动力学,从而保证了高倍率性能。DFT计算显示,1T和2H相界面处形成的固有电场吸引Na+朝向2H-MoS2,这加快了Na+的迁移速度。最近,设计了MXene-MoS2异质结构,目的是通过缩短扩散路径来显示Na+的更快传输。 这种稳定的异质结构进一步被认为能够带来连续的扩散通道,以实现更快的电子传递。合成的负极示出了0.2 A·g-1电流下出色的可逆容量315 mAh·g-1 和在1000个循环周期后2.0 A·g-1电流下的220.0 mAh·g-1。MXen显著降低了Na原子的吸附能量障碍(0.078 eV),这比MoS2@MoS2(1.28 eV)或MXene@MoS2(0.11 eV)等其他界面要低得多。MXene显著降低了MoS2的带隙以显著提高导电性,这些优秀的电化学性能的提高是由MXene保证的。
钒硫化物:钒硫化物(VS2)的电子传输效率比MoS2高得多,由于它们的金属性质,因此经常被认为是具有重要意义和适合作为负极材料的材料。VS2的理论研究表明,可以达到高达466 mAh·g-1的容量,输出电压低(0.48 V),其扩散系数可以达到与体相MoS2相比的七个数量级。负极的倍率性能可以直接通过Na+的扩散动力学来解释,结果表明,VS2中的Na+扩散动力学比MoS2(170 meV)低得多。此外,钠离子的活化能垒也比MoS2(780 eV)低。这些特性支持了使用VS2作为负极材料比MoS2有更多的优点的观点。然而,可用容量(≈250 mAh·g-1)比理论预测的要低得多,也比碳基材料低得多,这一直是使用它作为SIB电池负极的一个重要挑战。通过溶剂热法合成了具有层状结构的VS2纳米片,其在0.1 A·g-1下的容量为600 mAh·g-1,同时在循环保留性上表现出优异的性能(700个周期),在(2和5)A·g-1下分别为(83%和87%)。它也展示出优秀的倍率能力,在非常高的电流密度(20 A·g-1)下放电容量为277 mAh·g-1。这种卓越的电化学性能是由于赝电容机制,特别是在高电流密度下。花状VS2纳米片有利于电解液的渗透,足够增加的电解液界面使离子传输更快。值得注意的是,VS2纳米片提供了600 mAh·g-1的容量,这表明近2Na+参与到电化学反应中,这比其他参与硫酸亚铁电池的负极材料要多得多。通过氮掺杂,可以进一步改进这种花状VS2结构的电化学性能。例如,通过溶剂热过程在还原石墨烯上生长得到花状VS2/N掺杂碳(VS2/N-C)具有扩大的(001)面,然后进行碳化技术。合成的VS2/N-C@rGO纳米杂化物在1.0 A·g-1下提供了407 mAh·g-1的可逆容量,高倍率容量(8 A·g-1下的273 mAh·g-1),并且具有非常长的稳定性(1400个循环)。扩大的层间距(1.02 nm),显著降低了离子扩散壁垒,花状结构提供了数个活性位点,并且可以容纳在循环过程中的大体积膨胀,这两者共同改善了这个负极的电化学性能。
4.1.3 转移金属磷化物
由于其高理论容量和低电压,基于磷的材料已被探讨作为SIB的有希望的负极,但它们受到体积膨胀大和界面问题的困扰。在(脱)钠化过程中的粉化是磷基材料中容量衰减快的另一个原因,需要立即关注。Liu等人开发了一种通过红磷(Pred)浸渍碳纳米纤维(CNF)修复粉碎的策略。原位透射电子显微镜分析揭示,合成的红磷在钠化过程中变软,具有“液态般”的特性,可带来抗粉碎的延性和形变能力。浸渍策略进一步确保缓解副反应并改善电化学性能。Pred的存在有利于调节和适应较大体积变化率,通过纵向沿CNF内部空间膨胀。虽然在钠化过程中存在的刚性不能忽视,但主要的想法是尽可能减小这种刚性,以便容纳体积变化并限制粉碎。Pred的这种“液态般”的特性使容纳体积变化变得容易,并保护了电极的完整性。同时,也合成并测试了类似生物模仿的负极,如锡磷化物锚定在碳纳米管上,并作为SIB的负极。利用生物模仿的优势,该负极在0.2 C工作150个循环时,可提供742 mAh·g-1的容量,并在进行500个循环后,以2 C的更好倍率性能提供449 mAh·g-1的容量。在Sn4P3内部形成的单分散导电电子域会增加电化学活性表面积,最终能降低Rct。此外,P和Sn均匀分布在非晶碳基体内,这有助于电化学转化P,阻止可能在钠化过程中Sn的分离。至今,还合成了其他一些基于过渡金属的磷化物,并用作SIB的负极材料,如ZnP2, Zn3P2和NiP3/CNT等,并显示出令人印象深刻的结果。然而,必须进行密集的研究,以改进它们的化学性能,以提高SIB的整体电化学性能。
4.2 嵌入/插层型材料
当一个带电粒子嵌入到晶格内部时,一些宿主粒子必须离开或重排以接纳新的粒子。这需要结构处于一个位置具有良好的可氧化和还原性且具有良好的电子导电性的原子。因此,应该有一个大的空隙空间来容纳快速移动的离子,以提高存储系统的导电性。基于嵌入反应,已经鉴定并广泛研究了几种碳基和钛基化合物作为SIB的候选材料。特别是,碳基材料因其能够将Na+ 离子容纳在其结构中而受到更广泛的关注,尤其是硬碳,由于其相当大的容量(300 mAh·g-1)和低工作电压(接近零,约为0 V vs. Na+/Na)。最近,无序碳也被鉴定为Na+ 存储的宿主材料;然而,其存储/反应机制仍然是一个公开的争论。另一方面,廉价和较低的运行电位驱使研究人员将钛基化合物作为负极材料的另一途径。
4.2.1. 碳基材料
石墨碳:碳基材料是负极材料的首选,特别是石墨在LIBs中是商业化的负极。在嵌入过程中,Li+离子被嵌入到石墨层之间,并且通过相变在LIB中形成Li-石墨二元嵌入化合物(Li–GICs)。然而,当涉及到Na+嵌入时,这种现象明显不受欢迎,并且在电解质/电极中报告了显著的衰减。理论计算也表明,Na-GICs的能量并不如Li–GICs那样有利,,当一部分Na+嵌入时,石墨层会受到压力,这是由于二元Na-GICs的热动力学不稳定性所导致的,这种不稳定性源自Na和石墨层之间的尺寸不匹配。然而,关于尺寸不匹配是否是Na–GICs不稳定的唯一原因,或者是由于碱金属和C原子之间化学键的改变,人们的意见有所不同。这主要是由于在Na-石墨嵌入的第一阶段形成了不利的NaC6和NaC8,这些都在热力学上是不稳定的,通常是在SIB中使用石墨时容量差/限制的最明显原因。自从发现的溶剂化Na离子化学嵌入成功形成三元GICs之后,已经取得了一些进展,在二甘醇基电解质中,石墨在0.1 C下进行了1000个循环,容量达到≈100 mAh·g-1,这个意想不到的能力归因于diglyme溶剂化的Na(diglyme)2C20共插层入石墨。从这个初步成功中可以得到的想法是获得一个溶剂化的Na+ 离子并将其嵌入到石墨烯中。这种溶剂化可以通过具有非极性特性的高供体电解质得到,以成功地嵌入到天然石墨中。
还有一种方法是扩大石墨碳的层间间距。Wen等人成功地将石墨烯碳层扩展到0.43 nm。此扩展的石墨碳可以在20 mA·g-1下提供284 mAh·g-1的可逆容量,并在2000多个循环中表现稳定。通过电化学质谱研究发现,电解质分解在第一个循环被部分地观察到,而在随后的循环中被限制。TEM图像下没有SEI或破碎部分证实了这一点,此外,我们推测即使SEI存在,并且在第一个循环中由于溶剂共作用引起的显著体积膨胀而破裂,在随后的循环中也不会形成SEI,SEI的缺失促进了Na插入/提取过程中的电荷转移动力学。
非石墨碳:石墨烯:石墨烯是一种独特的二维碳材料,具有高化学/热稳定性和大比表面积等特殊性能,其优越的电子导电性使其成为SIB负极应用的理想材料。石墨烯可以提供更快的扩散路径,并且其大的比表面积可以提供更多的Na+/Li+嵌入通道。因此,石墨烯在作为EES负极材料方面具有巨大的潜力。此外,石墨烯可以还原为还原氧化石墨烯(rGO),进一步提高导电性和能量存储能力。Kim小组使用改进的Hummers法合成了rGO,在0.1C下实现了50个循环的逆向容量272 mAh·g-1,但在高电流密度下容量严重损失,与其高电性能形成对比,可能是因为在石墨烯存在的情况下电容有所损失,这是由于rGO片层在循环过程中倾向于团聚,削弱了其性能。然而,这个问题可以通过超声波技术、机械或热方法、或引入碳纳米管(CNT)来解决,从而创建无序结构以储存更多的Na+。最近,通过水热法合成了表面改性的rGO气凝胶,其层叠无序。在乙二胺和α-环糊精(CD)的存在下,表面改性的rGO显示出改进的电化学性能。在rGO-CD存在下,它在初始周期为0.1C时提供了约175 mAh·g-1的能量,并且在1C下经过100个循环保持了高倍率容量约60 mAh·g-1。尽管循环容量没有显著提高,但倍率性能得到了显著改进,这是由于rGO组装层的无序结构和引入的结构异质性。此外,缺陷的引入可以进一步改进电化学性能。例如,Daryabari等人通过计算研究证明,石墨烯量子点(QDs)的缺陷可以显著提高负极性能。石墨烯QDs中的缺陷可以改变其电子结构,并可以作为吸引/吸附Na原子的良好沉积点,显著改善了Na+储存性能。对于Na原子/离子来说,有缺陷的结构的吸附能通常是负的,这是防止Na原子团簇形成的理想因素。尽管负吸附能是获得高理论容量的理想状态,但由于缺陷的陷阱效应,它可能大大削弱电池中的迁移能力。因此,通常选择优化的吸附能量是更好的选择。
非石墨烯碳:硬碳:最先由Doeff等人报告的硬碳(HC),是SIB的一个先进负极材料,可以通过石油焦的热解得到。基于硬碳的实用全电池展示出高能量密度和耐久性,预期会引起对电网规模EES应用的兴趣。深入分析硬碳作为SIB负极材料的适用性之前,建立选择硬碳的理由是至关重要的。石墨和非晶碳是作为研究电池的负极广泛使用的两种碳质材料。此外,非晶碳(根据其石墨化能力)进一步被细分为软碳和硬碳。软碳在3000°C的温度下能够石墨化,而这个温度下,硬碳很难石墨化。值得在这里提到的是,石墨化程度取决于前驱体的选择,焙烧温度,升温速率,目标温度和退火处理条件。软碳的前驱体,如沥青,煤焦油和乙烯聚合物,可以在≈400°C的热解温度下经历相变进入流体样态。随后,随着进一步的热处理,他们转化为固态相的碳质材料。这种液体或焦油样中间物起着关键的作用,它促进了分子的重新排列,因而在软碳中促进了高程度的石墨化。而硬碳的前驱体在整个热解过程中保持固态,从而不可能高程度石墨化,这主要是因为前驱体要么高度交联,要么在初始热解阶段有发生交联的潜力。尽管存在一些误解,钠储存现象也没有明确的机制,但自2000年首次提出储存钠的发现以来,已经取得了一些进展,硬碳/无序碳中Na+的嵌入经过两个明确的阶段,由Dahn and Stevens提出的,众所周知的机制——“纸牌屋”来控制。第一阶段是斜坡区,对应Na+嵌入到无序石墨烯片中,而第二阶段(平台区)表示Na+对无序石墨烯的孔隙填充。最近,Ding等人提出这种观点,他们证明了HC在平台区的层间扩大与Na+嵌入有关,而不是孔隙填充,如最初提出的那样。很多团队支持这种观点,新的发现对这种机制提供了完全相反的观点。Bao小组合成了由海带得来的HC(KHC),具有扩大层间间隙范围(3.9-4.3)纳米。这种KHC在300个循环之后,200 mA·g-1电流密度下具有205 mAh·g-1的可逆容量以及1000 mA·g-1下的96 mAh·g-1的高倍率能力。调整形貌是增加初始容量的另一种策略,因为它导致电荷传递性更好,比表面积增大和稳定性增强。最近,有报告称商业硬碳(CHC)表面氧化显著提升了其电化学性能。通过化学处理策略,得到了具有可控氧官能团的扩展层间碳。改性CHC在20 mA·g-1下表现出341 mAh·g-1的可逆容量,远高于原始硬碳(270 mAh·g-1),在500 mA·g-1下表现出50 mAh·g-1的优异倍率性能。这种增强的电化学性能归因于氧官能团的引入,这带来了两方面的好处:一是通过扩大层间间距加速了Na+的扩散,二是增强了对Na+的吸附能力。
调整硬碳的孔隙度是改善Na+存储的另一种策略。为了使硬碳作为负极材料,必须充分理解孔隙度的变化。硬碳的处理通常通过机械化学处理来实现,这可以显著改变孔径,从而反映在其电化学性能的变化中。小角X射线散射和物理吸附显示,球磨过程中孔隙度发生变化,孔隙度的显著增加是不适宜的,而减少或封闭孔则适合Na+存储。这一点体现在原始硬碳的容量存储为58.5 mAh·g-1,而在球磨5小时后(孔隙体积增加),其容量仅为35.5 mAh·g-1。这一结果表明,高容量不仅仅是通过连续球磨增大孔径来实现的,还需要考虑其他方面,如层间距、掺杂和缺陷空位。Li等人提出,恒流充放电曲线中的斜坡区容量与硬碳的结构缺陷之间存在结构-性能关系。为了证明这一假设,作者通过微波处理650°C热解纤维素得到的碳,获得了缺陷硬碳。经过6秒的微波处理后,可逆容量从204 mAh·g-1增加到308 mAh·g-1,远高于1100°C退火7小时的硬碳(274 mAh·g-1)。这种显著的容量增加显然是由于微波和其高效能处理,保持了硬碳中大量的结构空位。这种缺陷结构允许斜坡区容量远高于传统硬碳。此外,微波加热是细调硬碳孔径以用于钠离子电池的极好工具。
这些讨论表明,目前对于硬碳中的钠嵌入过程是两步还是三步仍无定论,这仍然是一个开放的讨论领域。因此,进一步的理论和实验研究是必要的,以明确硬碳在钠离子电池商业化负极中的发展机制。尽管努力寻求单方面的机制共识,硬碳已成为应用最广泛的负极材料,显著提升了钠离子电池(SIB)的电化学性能。然而,实现硬碳商业化仍面临若干实际挑战。在前一部分讨论的多项挑战中,必须深入理解以下几个关键缺陷:平台区容量衰减、高活化能和低初始库仑效率(ICE)。平台容量衰减指的是在循环或长期使用中容量逐渐下降。硬碳负极的平台容量衰减可归因于以下因素:
(1)结构退化:钠离子的反复嵌入和脱出导致硬碳材料的结构变化和非晶化,导致容量损失。
(2)副反应:电解质和负极表面之间可能发生不必要的副反应,形成电活性较低的SEI层。这层SEI层阻碍了钠离子进入碳材料,降低了有效容量。
(3)不可逆反应:部分钠离子可能与碳结构发生不可逆反应,导致容量损失。
相比之下,高活化能指的是化学反应发生所需克服的能量障碍。在SIB中,高活化能通常指钠离子在硬碳负极中嵌入和脱出的能量障碍。与硬碳中的(去)钠化过程相关的高活化能会导致反应动力学变慢,影响电池的整体性能。这可能导致充放电倍率降低,难以实现高功率密度。高活化能可能归因于以下几个因素:
(1)有限的扩散路径:钠离子在硬碳中的嵌入和脱出需要通过材料的孔隙结构扩散。如果孔径或连通性有限,可能会增加离子传输所需的活化能。
(2)结构约束:刚性的碳结构可能对材料在循环过程中的膨胀和收缩施加限制,导致离子嵌入/脱出的能量壁垒增大。
(3)电解质限制:电解质的选择及其与硬碳负极的兼容性会影响活化能。高粘度或浸润性差的电解质可能阻碍离子传输并增加活化能。
研究人员正在探索各种方法以降低活化能,如优化碳材料的结构、修改电解质成分和引入纳米结构碳架构以改善离子扩散动力学。
本节还强调了硬碳的另一个关键方面:低初始库仑效率(ICE)。ICE是电池研究中的一个可量化指标,用于准确预测电池的寿命、能量密度和倍率性能。低ICE是硬碳在商业SIB中的实际应用瓶颈之一。在电池的第一个循环中,电解质会分解并在负极表面形成SEI层,导致不可逆的钠消耗。因此,硬碳表现出相对较低的ICE。然而,实验室测试通常使用半电池系统评估电极材料。在这种体系中,钠金属通常作为对电极,提供充足的钠离子。在这种情况下,即使负极在初始循环中消耗了大量的钠离子,仍有充足的钠量保证后续循环的进行。尽管这一因素在半电池系统中似乎无关紧要,但在全电池中却至关重要,因为正极材料提供了所有不可逆的钠离子。需要特别强调的是,许多研究人员在半电池测试中显示出高ICE,而这种高/低库仑效率只有在全电池配置中才有意义。因此,在制造过程中必须严格控制所有组分,以最大限度地提高电池的能量密度。在商业电池中,为防止充电期间锂金属析出并确保电池安全,通常保持负极/正极比为1.10-1.15,这实际上意味着负极设计的容量超出10-15%。各种研究小组提出了提高ICE的技术,其他综述文章中已提及,感兴趣的读者可参考相关文献。预锂化技术显著提高了锂离子电池(LIB)的ICE,并被认为在SIB中也可行,通过补偿不可逆的钠损失。实验研究表明,预锂化处理的硬碳(LPHC)在TEGDME基电解液中表现出优异性能。TEGDME中的LPHC显示出超过92%的ICE和220 mAh·g-1的比容量,远高于碳酸酯基电解液中的100 mAh·g-1。令人瞩目的是,即使在4C倍率下循环1000次后,其容量保持率(150 mAh·g-1)也比在碳酸酯基中的10 mAh·g-1高出约15倍。当制成Na3V2(PO4)3-LPHC全电池时,在TEGDME中的容量接近理论容量(约98 mAh g-1),远高于未经处理的硬碳(40 mAh·g-1)。这些有趣的电化学性能差异源于在TEGDME和碳酸酯基电解液中形成的不同SEI层。在碳酸酯基电解液中形成的SEI层非常厚,可能会消耗电极中大部分锂。核磁共振波谱数据表明,TEGDME中的相对强度峰比碳酸盐光谱中的强,这表明在LPHC中钠嵌入更容易。
除了上述讨论外,还必须深入理解几个其他重要参数,例如HC的容量极大地依赖于孔体积控制和比表面积。此外,选择合适的添加剂、电解质和粘结剂也是必须考虑的方面。我们还强调,必须进行更多的实验研究,以探索其他电解质中的潜在化学反应,从而建立SEI与HC电化学性能之间的关系。除此之外,还应借助计算工作,如从头计算分子动力学(MD)、机器学习(ML)和密度泛函理论(DFT)计算,以丰富我们的理解,并补充实验发现,从而为实验结论和商业化道路建立信任。
掺杂:掺杂是另一种调整电子结构和调控能源材料电化学性能的策略。当异质原子如硫(S)、氮(N)、硼(B)、磷(P)掺入碳材料时,会在材料中创建缺陷位点,从而吸附更多的钠离子,显著改善电极与电解质之间的相互作用。例如,氮掺杂的碳材料显著提高了离子传输能力,通过表面吸附和法拉第反应增加了比容量。Xu等人合成了氮掺杂的分级多孔碳微球(NHPCs),并将其用作负极材料。NHPCs在1000 mA·g-1的高电流密度下,经过200次循环后,展现出204 mAh·g-1的可逆容量,且倍率能力优异,达到256 mAh·g-1。在另一种方法中,硫掺杂在大孔催化碳纳米模板(S-MC-CNTs)中,表现出超过2400次循环的卓越循环稳定性和约99.8%的库仑效率。如此长期稳定性的本质在于硫掺杂,保证了硫掺杂碳边缘位点的化学/电化学稳定性。MC-CNTs中的碳是不饱和的,因为边缘碳只有两个键而不是sp2杂化所需的三个键,而硫终止的S-MC-CNTs则热力学稳定。负极材料的倍率性能可以通过磷掺杂进一步提高。通过一种新的溶液等离子体法合成了磷掺杂碳球,在100 A·g-1的极高电流密度下,表现出130 mAh·g-1的优异倍率能力,并在40000次循环后保持83 mAh·g-1的可逆容量。这种负极材料的几个优势包括:等离子体处理方法在材料中创造了大量的中孔和大孔,这些孔允许离子更快扩散;这些孔还能够容纳材料内部的体积变化,显著有助于结构稳定性,并提供大的电极/电解质界面以积累电荷。此外,首个循环后的EIS谱变化微小,表明形成的SEI层薄且稳固,抑制活性材料损失并提供更快的钠离子扩散,从而确保高倍率性能和增强的结构稳定性。
最近,还采用了双掺杂的理念,Lee等人通过机械化学路线合成了氮和硫双掺杂负极材料。由硫黑(SB)制得的氮和硫双掺杂多孔碳材料按化学计量比取用,球磨48小时得到SBG70-48 h。合成的负极在0.1 A·g-1时表现出282 mAh·g-1的倍率能力,并在200次循环后保持208 mAh·g-1的容量。该负极的电化学性能提升归因于双掺杂,它带来了多孔微结构和额外的活性位点以吸附钠离子,并促进更快的扩散动力学。经过对负极材料电化学性能改进的广泛讨论,现在我们已经知道,多尺度结构优化技术,如形貌工程、结晶度、层间距离增大和掺杂功能性改变了性能。最近,Wu等人提出了一种一步硼掺杂策略,带来了多尺度结构修改以改善电化学性能,作者利用氢键导向的共组装,然后在硼酸和葡萄糖(分别作为硼和碳源)的存在下进行高温碳化,得到硼掺杂的硬碳纳米球。具有BC3和B–C–O特征的合成负极在第二循环时表现出234 mAh·g-1的可逆容量,而无硼的表现为168 mAh·g-1 (相同电流密度为0.03 A·g-1)。它还表现出244.6 Wh·kg-1的高能量密度和超过200次循环的卓越循环稳定性。DFT计算表明,硼掺杂的BC3结构的HOMO–LUMO间隙(2.39 eV)远低于无硼的碳表面(2.66 eV)。由于碳材料中的电导率与带隙值直接相关,较低的带隙值表明BC3结构在碳表面上的引入促进了电子传输,加速了电化学扩散,并增强了钠离子存储。此外,BC3结构的吸附能(−2.10 eV)比无硼的碳表面的吸附能(−1.70 eV)低得多,这表明硼掺杂的碳表面及其边缘对钠离子存储具有更高的亲和力。
4.2.2 钛基材料
钛基能源材料因其低毒性、合适的工作电压、高安全性和低成本而成为负极应用的理想材料。值得注意的是,尖晶石型Li4Ti5O12(LTO)是一种广泛研究的材料,众所周知在电池研究中具有重要地位。这些化合物在钠电池中由Ti3+/4+的氧化还原电对驱动,,最近的研究集中在探讨新材料中的钠化/脱钠机制。
二氧化钛:二氧化钛有多种晶型,包括锐钛矿、金红石、板钛矿和青铜相TiO2,它们是广泛研究的钠离子电池负极材料。在这些多晶型中,锐钛矿TiO2因其Na+激活能垒与Li+相同而被广泛研究。Sun的研究小组通过溶剂热技术合成了无碳的锐钛矿/青铜TiO2微球(TiO2(A/B)–MS)。由于锐钛矿-青铜相界面结构的优势,合成的负极在不使用导电碳的情况下能够提供高比容量和功率密度。TiO2(A/B)–MS在0.1C时提供了221 mAh·g-1的容量,并在10C时表现出超过100次循环的优异稳定性。他们还发现,锐钛矿和青铜相的体相结构具有较高的迁移能垒(分别为0.56和2.2 eV),这是TiO2作为钠离子电池负极材料的主要缺点,而这种能垒在TiO2(A/B)–MS中得到了调节。TiO2(A/B)–MS中单个和多个Na+离子的界面结构的迁移能垒分别为0.49和0.30 eV,远低于其体相部分。令人惊讶的是,多Na+离子的离子扩散能垒低于0.19 eV,这进一步增强了TiO2(A/B)–MS作为高效钠离子电池负极候选材料的资格。最近研究发现,通过掺杂镍和锌可以改善青铜相TiO(TiO(B))的电化学储存性能。研究发现,掺杂5 at%的镍(Ni/Ti比为0.02/0.05/0.08)到TiO2(B)中成功地扩展4%的晶胞,并产生了缺陷,使能隙从3.28 eV减少到2.70 eV。相比之下,锌掺杂高度受限(Zn/Ti比0.02/0.05);尽管如此,它显著提高了导电性(3.29 × 10-9 S cm-1),而裸TiO2(B)的导电性为1.05 × 10-10 S cm-1。虽然锌掺杂在提高导电性方面有一些优点,但镍掺杂表现出最佳性能。在50 mA·g-1的电流密度下,镍、锌和裸TiO2(B)分别在100次循环后获得了(173, 151, 和 140)mAh·g-1的可逆容量。镍掺杂不仅改善了循环性能,还显著提高了倍率性能。几个因素有助于性能的提升,例如1)增加电子导电性,2)加快Na+动力学,3)在嵌入/脱出机制过程中增强结构稳定性。
缺陷工程是另一种显著改善过渡金属氧化物电化学性能的工具。Patra等人研究了用于钠离子电池负极的氢化相(H)金红石(TiO2-R)和锐钛矿(TiO2-A)。研究发现,H-TiO2-A在多个方面优于H-TiO2-R。例如,电子导电性按增加顺序为:TiO2-R < TiO2-A < H-TiO2-R < H-TiO2-A(分别为2.4 × 10-10, 2.3 × 10⁻⁸, 4.0 × 10⁻⁷, 和4.8 × 10⁻⁴ S·cm-1),而H-TiO2-A的比容量(100 mAh·g-1)在相同电流密度(10 A·g-1)下远高于H-TiO2-R(≈50 mAh·g-1)。钠嵌入能量按减少顺序为TiO2-R > H-TiO2-R > TiO2-A > H-TiO2-A(分别为0.5221, 0.1915, -0.0543, 和-0.3031 eV),这表明系列中的最后一个具有最佳的充放电特性。钠离子电池中的Rct(Ω)/DNa+ (cm2·s-1)按增加顺序为:TiO2-R < TiO2-A < H-TiO2-R < H-TiO2-A(分别为86/1.3 × 10-14, 74/3.1 × 10-14, 49/5.2 × 10-13, 和23/7.4 × 10-13)。H-TiO2-A由于锐钛矿-TiO2的二维扩散路径,具有最佳的值。要彻底了解H-TiO2-A优异性能的根本原因,了解氢化对晶体结构的影响是重要的。氢化后,表面处于无定形状态,而在苛刻的还原气氛中,H2可以从晶体中脱出氧原子,产生氧空位(OVs)。DFT计算显示,OVs促进Na+嵌入,因为H-TiO2-A的Eintercalated-OV(-0.3031 eV)优于H-TiO2-R(0.1915 eV)。此外,氧空位创建了更开放的结构以容纳更多的钠离子,提高了负极的循环性能和稳定性。
碱金属钛酸盐:具有尖晶石结构的钛酸锂(Li4Ti5O12)因其长寿命、较小的体积膨胀(也称为“零应变”材料)和高热力学稳定性(约1.556 V相对于Li+/Li的高电压平台)是一种很有前途的材料。Zukalova等人研究了纳米晶尖晶石(nano-LTS)和商业LTS中的钠离子嵌入反应机制,揭示了LTS结构在此过程中受到的应力导致其初始容量较低。拉曼光谱研究显示,Na+嵌入后,纳米LTS中形成了正交相Li0.5TiO2。纳米LTS和LTS的电化学容量分别为156和75 mAh·g-1,其中纳米LTS的容量接近理论计算值。然而,由于Na+嵌入引起的不可逆结构变化,纳米LTS在循环过程中显示出容量下降。需要提到的是,Li+嵌入遵循公认的两步过程,而钠离子电池中的Na+嵌入则有三个步骤。由于Li+和Na+的离子半径不同,Na+离子占据Li4Ti5O12晶格的16c位点,并通过以下反应引发两个岩盐相的LiNa6Ti5O12和Li7Ti5O12的形成:2Li4Ti5O12 + 6Na+ + 6e-↔ Li7Ti5O12 + Na6LiTi5O12。Kim的研究小组研究了铬掺杂对LTO低扩散系数和绝缘性的显著改善。优化后的Cr掺杂LTO在1C下经过400次循环后提供了110 mAh·g-1的可逆容量,在10C下的容量几乎是未掺杂LTO的三倍(75 mAh·g-1)。铬(Cr)是一种材料,其化学计量量(低掺杂和高掺杂)在电化学性能中起着非常重要的作用(详见后文)。作者还展示了低掺杂(L)和高掺杂(H)Cr的情况。有几项特性促进了适当掺杂的LTO的电化学性能提升。例如,轻掺杂Cr-LTO(L-C-LTO = 176.1 m2·g-1)的BET表面积远高于LTO(147.9 m2·g-1),也高于重掺杂Cr-LTO(H-C-LTO = 170.15 m2·g-1)。有趣的是,L-C-LTO具有高比表面积,这意味着它具有众多介孔结构。由多尺寸孔组成的介孔结构比单一尺寸孔具有更好的物理化学性质,这意味着L-C-LTO作为负极比其他材料具有优势。另外,从DFT计算得到的迁移势垒表明,Na+在Na6LiTi5O12和Na6Li0.5Ti4Cr1.5O12的八面体位点(16c)之间的势垒分别为0.84和1.03电子伏特。Cr掺杂LTO中较高的迁移势垒意味着在Cr掺杂后迁移受阻。这可以很容易地从Na6Li0.5Ti4Cr1.5O12中Cr-O(2.06 Å)的键长略小于Ti-O、Na-O和Li-O的2.10、2.32和2.18 Å得到理解。较短的键长减小了Na+迁移路径的尺寸,从而增加了迁移势垒。然而,L-C-LTO中测得的电导率(1.1 × 10-7 S·cm-1)高于(1.7 × 10-7S·cm-1),这表明更低的电荷传输阻力,更低的极化。此外,L-C-LTO的(DNa+)扩散系数(7.24 × 10-20 cm2·S-1)高于(1.1 × 10-20 cm2·S-1),这证实了适当的Cr掺杂提高了电导率。继续讨论为什么重度Cr掺杂对电化学性能有负面影响,最近已经确立,Cr3+在高电压下具有3e-氧化还原中心(Cr3+/Cr6+);然而,它形成了竞争的层状相,这显著限制和限制了其溶解度。在寻找其他钛酸盐的过程中,钠钛酸盐(NTO: Na2Ti3O7)是最受关注的适用于钠离子电池负极材料。由Naeyaert等人和Woo等人观察到,钠钛酸盐具有单斜晶体结构(M-Na4Ti5O12)和三方晶体结构(T-Na4Ti5O12)。Palacin的研究小组首次报道,Na2Ti3O7在低操作电位(0.3 V vs. Na+/Na)下可以吸收2 mol的Na+ /f.u.。理论计算报告称,在Na2Ti3O7结构中嵌入2 mol的Na+形成Na4Ti3O7,这产生强烈的静电排斥力,导致结构不稳定和低工作电位。此外,全钠化状态下的这种静电排斥力还诱发了一种自我松弛现象,通常有利于缓解应力。Sun的研究小组通过水热法合成了NTO纳米管,以100 mA·g-1的倍率可逆地提供了126.2 mAh·g-1的容量,并具有出色的循环性能。
合金化-去合金化是另一种从Sn、Bi等金属或IVA族金属元素以及VA族非金属元素中产生高比容量的机制。然而,取决于宿主材料和钠离子的钠化程度,Na+可以引起巨大的体积变化,可能导致晶体结构的崩溃。因此,需要进行广泛的研究来理解它们的机制,以从中获得最大的益处。例如,Si由于其对4.4 Li+/Si-原子的吸收能力,在锂离子电池中是理想的负极材料,但由于与Na+的吸收能力显著较低(1 Na+/Si),几乎无法在钠离子电池中使用。另一方面,非晶态Si钠化是可能的,Han等人提出最大的钠化二元相是1:1(NaSi),可以提供954 mAh·g-1的容量。他们发现,使用非晶态Si可以改善可逆钠化,但仍受到有限的Na+扩散动力学和原始Si嵌入过程中的正向结合能的限制。非晶态Si既可以避免晶态Si的大幅体积膨胀(约300%),又带来各向同性的应变/应力。此外,非晶态状态中的活化能障碍比晶态Si低得多(0.4电子伏特),有助于改善电化学性能。此外,3D多孔结构可以通过电解液的渗透和电子/离子路径的缩短进一步提升性能,但更重要的是可以在循环过程中容纳大幅体积膨胀而不损失电极的结构完整性。Younis等人理论设计了Si2BN的3D多孔纳米带状结构,展示了在低电压(0.15 V)下341.61 mAh·g-1的容量,体积膨胀率为2.7%。这种多孔结构具有直接的半导体特性,带隙为0.67电子伏特,迁移势垒较低(0.19电子伏特),显著有助于获得更好的电化学特性。
锗是另一种用作负极材料的材料,其化学性质与Si类似,如最多与一个Na结合。理论计算预测,Na可以与Ge合金化形成可提供接近370 mAh·g-1容量的Na-Ge合金。然而,与Si一样,Ge也遭受着无法以其晶体形式存储Na的困境,因为Na+的离子半径较大,导致在嵌入点之间跳跃的活化能较高(1.5电子伏特)。在Na64Ge64体系中进行的第一原理计算揭示,非晶态Ge(a-Ge64)与Na原子的混合比晶态Ge(c-Ge64)更容易。非晶态Ge中的钠扩散率约为4.876 × 10-9 cm2·s-1(300 K),比其他合金化化合物快一个数量级,同时活化能(0.709电子伏特)也降低了,可以提供理论容量达369 mAh·g-1。最近,设计了基于GeP的负极材料,其电化学性能优于Ge和Ge + P混合物。第一原理计算显示,沿[010]平面的活化能为0.28电子伏特在Na+中足够,而在Ge中要高得多(1.02电子伏特)。此外,嵌入NaxGeP的中间步骤具有金属特性,有助于提高电导率。有趣的是,GeP经历了一种自愈三阶段机制(嵌入、转化和合金化),由于形成能量低,保证了其层状结构的重建,并有助于提升电极的整体电化学性能。
通过这些综合研究进展的分析,文章展望了SIBs技术的未来发展方向,并对其在全球能源转型中的作用表达了乐观的预期。随着对SIBs的深入研究和技术创新,我们有理由相信,钠离子电池将在未来能源存储领域发挥更加重要的作用,为实现碳中和目标和构建清洁能源体系提供坚实的技术支撑。
让我们共同期待SIBs技术的进一步突破,为全球能源的可持续发展贡献力量。感谢您对本系列文章的关注,我们将继续深入探讨SIBs的最新动态和研究进展,敬请期待。
参考文献:A. N. Singh, M. Islam, A. Meena, M. Faizan, D. Han, C. Bathula, A. Hajibabaei, R. Anand, K.-W. Nam, Unleashing the Potential of Sodium-Ion Batteries: Current State and Future Directions for Sustainable Energy Storage. Adv. Funct. Mater. 2023, 33, 2304617.

联系我们| bd@xbpower.com.cn
公司网址|https://www.xbpower.com.cn
公司地址 | 北京市大兴区联东U谷·大兴高端智造产业园
