

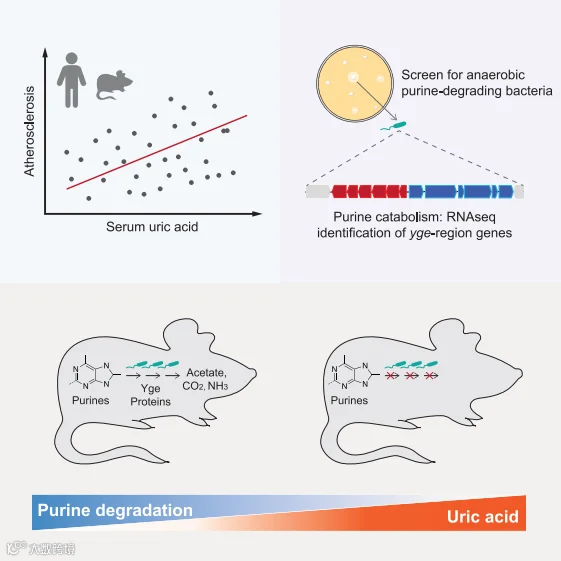
摘要
影响宿主炎症性疾病进展的微生物和微生物途径在很大程度上仍未确定。在这里,我们发现动脉粥样硬化负荷的变化部分是由肠道微生物群驱动的,并且与小鼠和人类的循环尿酸(UA)水平有关。我们确定了跨越多个门的肠道细菌分类群,包括杆菌门,梭杆菌门和假单胞菌门,它们使用多种嘌呤,包括UA作为碳和能量的厌氧来源。我们确定了一个基因簇,编码厌氧嘌呤降解的关键步骤,并广泛分布在肠道细菌中。此外,我们的研究表明,嘌呤降解细菌的定植会调节肠道和全身UA和其他嘌呤的水平。因此,肠道微生物是宿主嘌呤稳态和血清尿酸水平的重要驱动因素,肠道细菌嘌呤的分解代谢可能代表了肠道细菌影响健康的一种机制。
图a 嘌呤代谢示意图
引言
代谢紊乱包括肥胖、2型糖尿病和动脉粥样硬化,历史上被视为主要由过度摄入高热量食物引起的脂质疾病。然而,现在广泛认识到,慢性炎症在这些疾病的发展和进展中起着核心作用。动脉粥样硬化是心血管疾病(CVD)的主要原因,以血管炎症为特征,受多种遗传和环境因素的影响。在人类群体中进行的大规模全基因组分析已经确定了超过100个与动脉粥样硬化本身显著相关的位点,以及数百个与动脉粥样硬化相关的特征,如血浆脂质、肥胖和糖尿病相关的位点。然而,尽管遗传对动脉粥样硬化有显著影响,环境,特别是饮食,也在其进展中起着重要作用。此外,一些最近的研究提供了证据,表明饮食对疾病进展的贡献通常是通过肠道菌群介导的。
肠道菌群对代谢和炎症性疾病有着深刻的影响。饮食和宿主来源的因素调节肠道菌群的组成,而肠道菌群又转化宿主摄入的饮食成分,产生与免疫系统和几乎所有宿主器官,包括血管系统,相互作用的生物活性分子。过去一个世纪以来,食品生产、饮食习惯、抗生素使用和生活方式的变化导致了肠道菌群的重大变化,并以不协调的方式影响了人类健康:急性传染病的发病率下降,而慢性炎症性疾病的发病率上升。
此外,一些由饮食衍生的肠道细菌产生的代谢产物被发现是代谢和心血管疾病的潜在驱动因素。这些代谢产物构成了环境暴露和宿主细胞功能之间的直接联系,包括几种尿毒素(即肾功能受损的受试者不能正确排除的废物),包括对甲酚硫酸盐、吲哚硫酸盐、三甲胺-N-氧化物(TMAO)和苯乙酰谷氨酰胺。例如,膳食胆碱、甜菜碱和肉碱作为TMAO的底物,TMAO是由肝脏从肠道细菌产生的三甲胺(TMA)生成的。TMAO增强了小鼠的炎症和主动脉血栓形成,并且与人类的CVD风险相关。最近发现,由菌群从膳食蛋白质合成的苯乙酰谷氨酰胺,通过宿主的G蛋白偶联受体增强了血小板激活和血栓形成。这些证据支持了肠道细菌代谢通过调节循环中尿毒素的丰度,对CVD相关特征的贡献的观点。
几种嘌呤,包括黄嘌呤、次黄嘌呤和尿酸(UA),也被认为是尿毒素,并且导致慢性肾脏病患者观察到的几种症状。UA是人类嘌呤代谢分解的终产物,主要是因为它在浓度达到饱和水平时引起的并发症而被研究,形成促炎症的晶体沉积在关节(例如,痛风)。然而,一项最近的研究表明,UA在溶解度范围内的浓度可以通过诱导AMP激活的蛋白激酶(AMPK)介导的炎症促进动脉粥样硬化。UA加剧了炎症、内皮功能障碍,增加了肾素-血管紧张素-醛固酮系统的活性,并且在高血压和心力衰竭的患者中增加。
一些研究表明,对高尿酸血症患者有效降低UA产生或增加其排泄的药物干预可以改善心肾结局。虽然肾脏在调节循环中UA的水平方面起着重要作用,但这种代谢产物的相当一部分被分泌到肠道,并且一项最近的宏基因组学研究确定了与血液中UA水平相关的细菌途径。然而,尚未建立这种尿毒素的丰度与特定肠道细菌之间的因果关系,尽管已经从生化角度研究了厌氧嘌呤代谢的细菌机制,但其遗传基础仍未明确。
我们试图研究肠道菌群在动脉粥样硬化中的作用,并确定可能对疾病负担有贡献的微生物途径。首先,我们将来自具有不同动脉粥样硬化表型的小鼠品系的微生物群落移植到无菌(GF)载脂蛋白E敲除(ApoE KO)小鼠中。我们发现,微生物驱动的动脉粥样硬化进展的变异与嘌呤代谢产物(包括UA)的丰度相关。我们还观察到,这种促炎症的代谢产物与人群中的动脉粥样硬化负担和肠道微生物特征相关。
我们鉴定了能够厌氧降解嘌呤的细菌类群,发现了一个编码厌氧嘌呤降解所需关键组分的基因簇,证明了影响其活性的环境因素,并显示了含有该位点的类群的定殖降低了肠道中的多种嘌呤和全身性的UA水平。总之,这项工作加强了肠道微生物和动脉粥样硬化之间的联系,并提供了细菌代谢如何影响宿主生物学的见解。
结果
1、肠道微生物调节动脉粥样硬化的进展以及与小鼠疾病相关的血浆代谢物。
以前的工作揭示了来自杂交小鼠多样性面板(HMDP)的100个近交小鼠品系中动脉粥样硬化负担的巨大差异,这些小鼠品系具有不同的微生物群落。我们假设肠道菌群对HMDP品系中观察到的疾病进展的变异有贡献。我们将来自四个HMDP品系的盲肠样本移植到GF ApoE KO受体小鼠中。我们选择了两个表现出大的动脉粥样硬化病变(AXB10/PgnJ和BXD5/TyJ)和两个表现出很少疾病迹象(BTBR T+tf/J和BXA8/PgnJ)的品系,以下分别简称为“AXB10”、“BXD5”、“BTBR”和“BXA8”。移植后的小鼠在添加了0.2%胆固醇的饲料上饲养了8周。在此期间,评估了动脉粥样硬化病变、肠道菌群组成和疾病生物标志物(图1A和S1)。
我们发现,与来自HMDP供体的盲肠群落定殖的小鼠相比,来自倾向于动脉粥样硬化发展(即AXB10和BXD5)的供体的盲肠群落定殖的小鼠表现出更大的病变。这些结果支持了肠道菌群对动脉粥样硬化的发展有贡献,并可能对HMDP品系中观察到的疾病负担的变异有贡献的观点(图1B-1G)。传统的CVD危险因素,如体重和胆固醇,以及先前确定的肠道菌群衍生的代谢产物,包括内毒素(LPS)、TMAO和短链脂肪酸,都不能解释移植小鼠之间观察到的动脉粥样硬化负担的差异(图S1)。
通过对移植小鼠的盲肠内容进行宏基因组学分析,鉴定了1,649个功能特征和52个细菌分类特征。功能特征的主成分分析显示,按供体品系分组(p值<0.001,PERMANOVA),表明每个使用的四个HMDP品系具有独特的肠道细菌功能特征(图1H)。Pearson相关性分析确定了一些与受体小鼠动脉粥样硬化病变大小相关的细菌功能(图S2A和S2B)。这些包括与嘌呤的产生和转化相关的几个功能。我们还观察到,包括能量代谢和氨基酸代谢在内的细菌途径在与动脉粥样硬化病变大小呈正相关的功能中富集。
为了进一步研究微生物移植是否影响了与疾病相关的循环代谢物,我们使用超高效液相色谱(uHPLC)-串联质谱(MS/MS)对血浆样品进行了代谢组分析。共测定了682种代谢物(表S1)。Pearson相关性分析确定了嘌呤代谢物,包括黄嘌呤、黄嘌呤苷、肌苷和UA,与动脉粥样硬化病变大小呈正相关(图1I和1J)。总之,这些结果可能表明,肠道微生物影响了移植小鼠的动脉粥样硬化进展和血液中嘌呤的丰度。
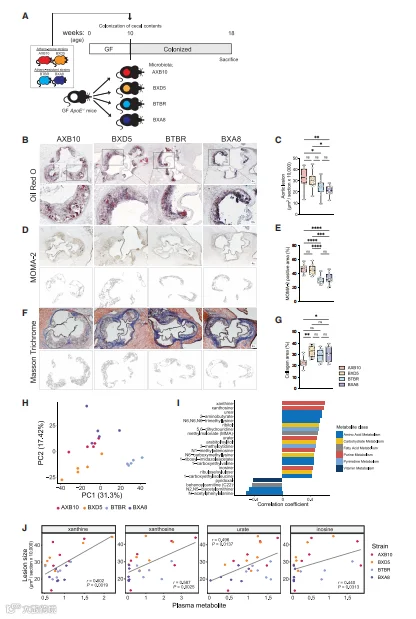
图1 在移植的GF ApoE KO小鼠中,血浆嘌呤水平与动脉粥样硬化程度有关。
2、人体中的血清UA与肠道微生物特征和亚临床动脉粥样硬化相关。
UA是人类嘌呤代谢的终产物,已经证明它能够引起炎症,诱导内皮功能障碍,并刺激平滑肌细胞增殖。我们探索了动脉粥样硬化、肠道细菌和UA在先前对肠道菌群和葡萄糖稳态进行了表征的人群(n = 998)中的关联。冠状动脉钙化(CAC)评分测量用于评估疾病负担。
动脉钙化是估计动脉粥样硬化斑块负担的公认代理指标。我们首先根据他们的CAC评分状态对个体进行分类:CAC评分= 0(即没有可检测的血管钙化,n = 492)与CAC> 0(n = 497)。逻辑回归分析显示,UA浓度的分布在这两组个体之间有显著差异(图2A),CAC> 0的个体显示出更高的UA平均水平和中位数水平。
此外,对CAC评分> 0的个体进行的Perason相关性分析显示,UA和CAC评分之间存在显著的正相关(rho系数= 0.14,p值< 0.001,图2B)。然后,我们应用极端梯度提升(XGBoost)回归来识别与UA水平相关的肠道细菌类群。
XGBoost是一种基于决策树的集成机器学习算法,使用梯度提升方法。在混合线性回归模型中校正了协变量(CAC评分、体重指数、性别、甘油三酯和糖化血红蛋白)后,与UA水平相关的前10个特征显示在图2C和表S2中。有趣的是,我们发现了多个属于梭菌纲的类群,它们与UA水平呈负相关。总之,这些结果表明,肠道菌群,特别是梭菌纲内的类群,可能影响UA水平。这些数据也与上述无菌小鼠的工作和先前将UA与人类CVD联系起来的工作一致。
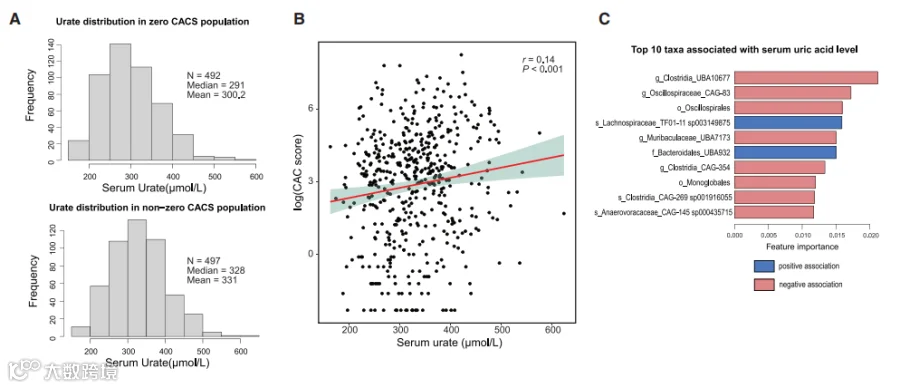
图2. 人体血浆UA水平与冠状动脉钙化(CAC)评分呈正相关
3、肠道微生物组调节盲肠和代谢循环中的嘌呤丰度
我们接下来研究了肠道菌群是否调节了肠道和循环中嘌呤的丰度。我们使用液相色谱-串联质谱(LC-MS/MS)在无菌(GF)小鼠和常规饲养(Conv)动物的盲肠内容物和血浆中定量测定了嘌呤相关的代谢物,包括核苷酸、核苷和核苷碱(表S3A和S3B)。我们发现,与Conv小鼠相比,GF小鼠的盲肠内容物中大多数嘌呤都降低了,少数例外是GF小鼠中增加了,特别是UA和尿囊素,这两者都是小鼠的终末嘌呤代谢物(图3A和3B)。考虑到盲肠/粪便中的嘌呤来源于饮食类型、细菌和宿主的周转和代谢,这些分析不能确定这些化合物在肠道中的来源和命运。
盲肠嘌呤的偏最小二乘判别分析(PLS-DA)显示了两组在主成分1和2上的分离。虽然血浆样品的分离不太明显(图S3A和S3B),但我们发现GF小鼠的血浆中UA水平显著高于Conv小鼠(图S3A和S3C)。这一结果用酶法测定UA的方法得到了证实(图3C)。这些结果再次表明,肠道菌群调节了肠道和全身的嘌呤的丰度,并促使了试图分离厌氧嘌呤降解细菌(PDB)的尝试。
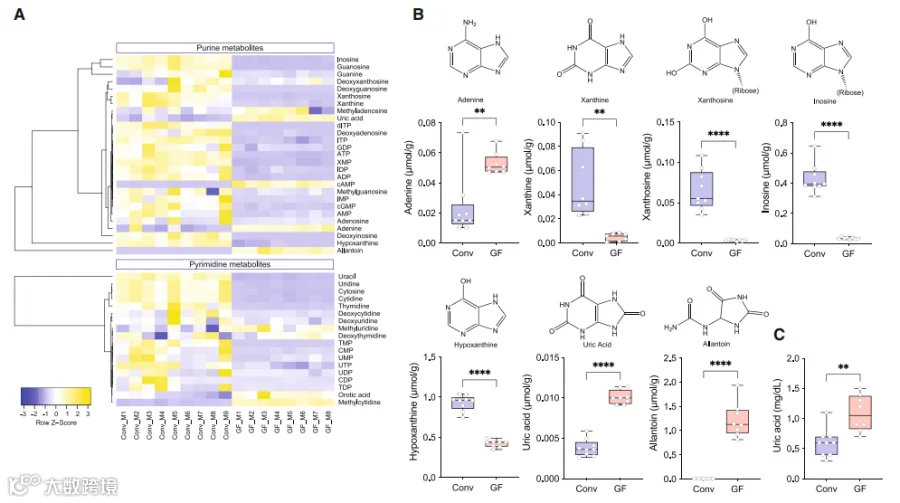
图3. 肠道微生物组调节盲肠和代谢循环中的嘌呤
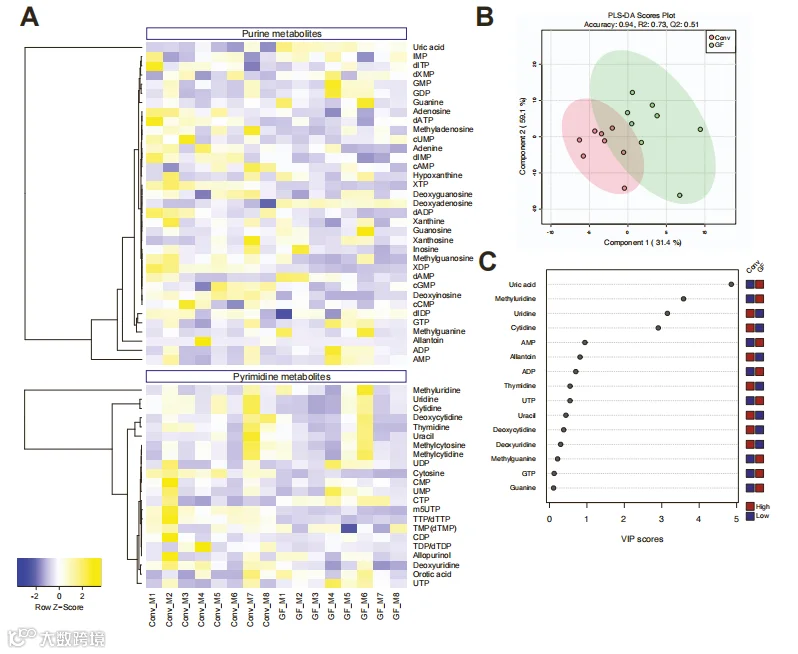
图b:Conv和GF小鼠血浆样品中嘌呤代谢产物的靶向定量
4、人类肠道细菌在厌氧条件下降解嘌呤。
假设肠道细菌通过将嘌呤代谢为非嘌呤产物来影响嘌呤水平,但尚未鉴定出从人类或小鼠肠道中分离的能够在这些代谢产物上厌氧生长的菌株。我们尝试在补充UA作为主要碳和能量来源的培养基上使用粪便浆(人类样本)进行厌氧富集。在带有顶部琼脂层的双层琼脂板上接种富集物并分离菌落,所述琼脂层提供饱和量的UA或其他嘌呤,验证了E.bolteae ATCC BAA-613种型菌株的生长和UA利用率,该菌株先前已测序。我们观察到,在24小时内,该菌株在补充了12 mg UA的10 ml培养物中降解了49.6 μmol的底物,积累了106.9 μmol乙酸盐,这是该生物体的已知发酵产物。并通过使用上述方法筛选了34个分离株的培养物,其中包括来自六个门的肠道细菌。
通过应用细菌贴片的生长和不溶性嘌呤底物的消失,证明了这些菌株在厌氧条件下对尿囊素和嘌呤依赖性的生长特征。筛选显示了在杆菌门(厚壁菌门)、梭杆菌门和假单胞菌门(变形菌门)中嘌呤利用的证据,尽管这种特性在属于这些门的菌株中并不普遍。特别值得注意的是,这些测定显示了UA降解菌株之间不同的嘌呤利用能力,但六种拟杆菌菌株中没有一种在UA、尿囊素或腺嘌呤上显示出任何生长。
总之,上述结果表明,普通肠道细菌可以利用嘌呤获取碳和能量,其他碳源和金属的可用性可以调节这一过程。
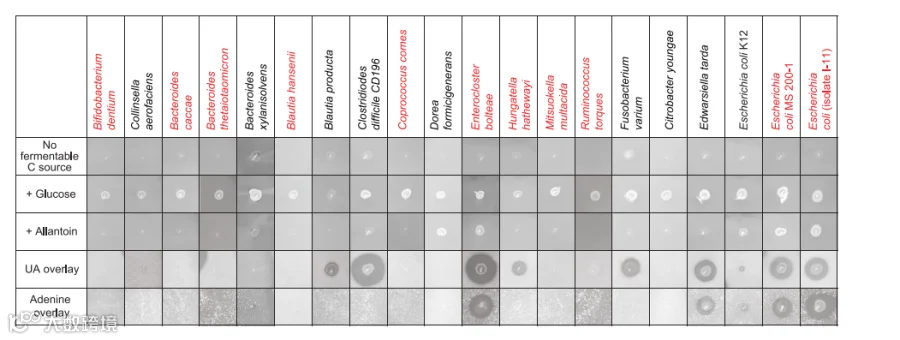
图4. 肠道细菌分离株使用嘌呤作为碳和能源
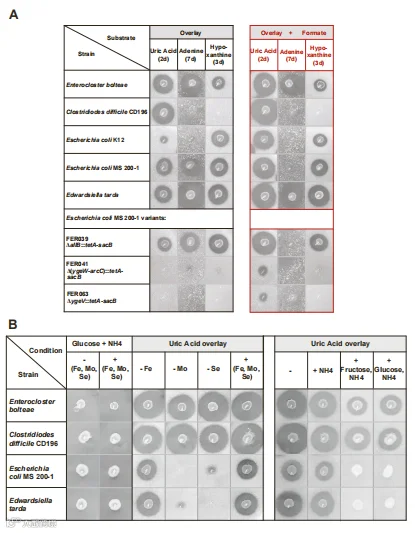
图C 环境因素影响嘌呤利用
5、PDB调节盲肠和代谢循环中嘌呤的丰度。
为了在体内测试上述PDB的影响,我们创建了降解UA能力不同的合成细菌群落,并将其用于GF小鼠的定植。用一个核心群落定植GF小鼠,该核心群落包括跨越人类肠道主要门的七个物种,且该群落在体外不降解嘌呤。这些群落中使用的每种菌株的UA利用率的体外测试如图所示(以红色突出显示),测量粪便移植的组合活性验证了用PDB定殖是UA降解所必需的。第三组动物在整个实验过程中保持GF。通过COPRO-seq(测序群落分析)分析移植的细菌群落,这些群落中的所有分类群都成功地在GF小鼠的肠道中定植,E. bolteae在三种PDB中含量最高。接下来,我们对盲肠内容物和血浆中的嘌呤、嘧啶和相关代谢产物进行了靶向定量。数据的整体分析显示,GF小鼠和“核心”或“核心加PDB”小鼠的盲肠嘌呤相关代谢产物有不同的模式,其中GF小鼠盲肠中的核苷增加,但核苷酸减少。
令人惊讶的是,与用核心群落定殖的小鼠相比,用核心加PDB群落定殖的小鼠在盲肠中显示出显著更高的UA水平,而在用三种PDB共同定植的小鼠中,包括次黄嘌呤和尿囊素在内的其他嘌呤/核苷的盲肠水平显著降低。
值得注意的是,与UA相比,在小鼠肠道中检测到的这些代谢物浓度明显更高。PLS-DA分析将核心血浆样本加PDB与其他两组的血浆样本分离,核心加PDB的定殖导致血浆中几种嘌呤代谢产物的水平持续降低,包括UA。血浆UA结果再次通过酶测定得到证实。
总之,这些结果表明PDB影响肠道中几种嘌呤的水平,特别是局部和循环中UA的水平。
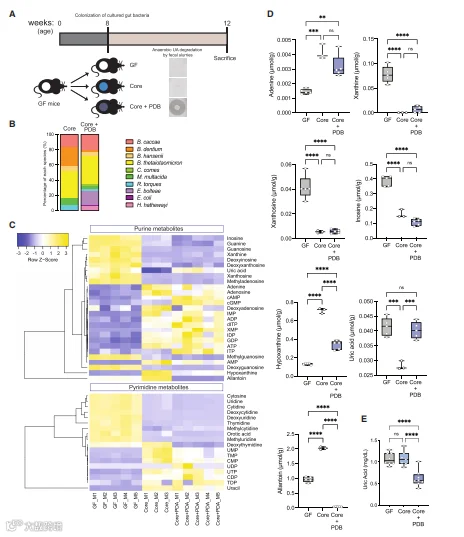
图5. 嘌呤降解菌(PDB)调节盲肠和代谢循环中嘌呤的丰度
6、转录分析鉴定多种嘌呤厌氧生长所需的细菌基因。
在系统地确定了PDB在降低UA水平方面的作用,并鉴定了能够厌氧代谢嘌呤的肠道分离株后,我们试图鉴定编码这些功能的基因。在补充有UA或木糖加NH4Cl(此后称为“木糖”)的培养基中培养E.bolteae的培养物。对于这两种底物,收获对数期细胞,并对文库进行测序。根据两种底物上生长的相对表达水平绘制标准化为基因大小的百万分之读数(RPM),仅限于3217(5993中)个差异表达基因(FDR<0.01)。正如预期的那样,编码30S和50S RNA聚合酶(RNAP)亚基的基因显示出轻微的偏向木糖底物侧,这与在该底物上观察到的更快的生长速率和RNAP亚基表达的限速性质一致。相对于木糖,UA上的生长促进了51个基因的更高表达,包括几个预测编码微量营养素转运功能,一个甘氨酸裂解系统,以及一个可能的电子分叉氢化酶。在UA上生长的E.bolteae中,有两个相邻且方向不同的假定操纵子,每个操纵子编码6个基因,占所有RNA-seq读数的12%。图中高度上调的基因由蓝色和红色圆圈表示,并绘制了相应的操纵子和假定基因产物图。在所有分类群中,嘌呤发酵生物的保守染色体区域的比对表明了五个基因(大肠杆菌命名法:dpaL、hydA、ssnA、ygeY和xdhD),尽管它们既不具有保守的组织,也不完全存在于跨门的连续基因组区域中。带有ygeW-dpaL-ygeY-hydA-arcC操纵子缺失的 E. coli MS 200-1变体作为野生型菌株在提供葡萄糖或尿囊素的培养基中生长,但不能使用UA、腺嘌呤或次黄嘌呤作为碳和能量源厌氧生长。相反,携带allB缺失的E. coli MS 200-1变体,编码催化厌氧尿囊素代谢第一步的酶,不能利用尿囊素,但保留了分解代谢UA、腺嘌呤和次黄嘌呤的能力,表明该生物体中尿囊素和嘌呤代谢的不同机制,并且与UA生长的E.bolteae细胞中所有基因的低表达水平非常一致。最后,携带编码单组分sigma 54型转录因子的ygeV基因缺失的E. coli MS200-1变体在葡萄糖和尿囊素上正常生长,但在所有测试的嘌呤上都未能生长,这与先前表明YgeV对相邻ygeW操纵子的转录调节的数据一致。为了评估这种细菌基因簇在体内嘌呤代谢中的作用,我们使用了上述相同方法,其中GF小鼠用缺乏PDB的核心群落、“核心群落加E. coli MS 200-1野生型”或“核心群落加缺失变体FER041”定植。在测试条件下,野生型和变体在肠道中的定殖水平相当。与用核心或缺失变体FER041定植的小鼠相比,用野生型菌株定植导致血浆UA水平较低。
总之,这些结果表明,这些基因编码的细菌功能有助于体内UA稳态。
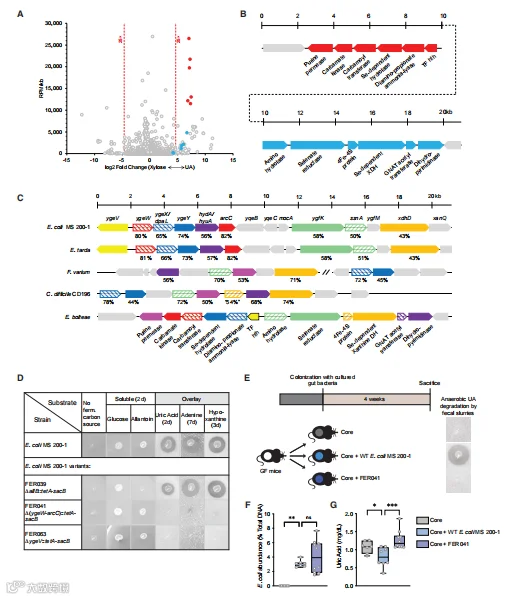
图6. 利用嘌呤生长的厌氧细菌所需基因簇的鉴定
7、细菌基因组和移植小鼠嘌呤降解功能基因的检测。
在小鼠中确定了无氧嘌呤代谢和UA稳态所需的基因后,我们试图确定含有这些基因的细菌类群。使用STAR方法中描述的参数对NCBI RefSeq基因组数据库(RefSeq_genomes)进行了BLASTP。检测到230个非冗余细菌类群,这些类群具有在所有实验确认的嘌呤降解类群(dpaL、hydA、ssnA、ygeY和xdhD)中可靠检测到的五个基因。最后,评估了ApoE无菌小鼠盲肠中这些基因的丰度,并将它们的丰度与盲肠中定量的嘌呤相关代谢物水平相关联,发现盲肠中几种嘌呤相关代谢产物(包括脱氧黄嘌呤、黄嘌呤和UA)的水平与参与厌氧嘌呤降解的基因丰度呈负相关。总之,这些结果突出了这些基因作为肠道嘌呤分解生物标志物的潜力。了解如何操纵肠道微生物群中嘌呤消耗物种的表现和功能(而不是相关基因的丰度),可能会为预防或治疗高尿酸血症和相关疾病提供方法。
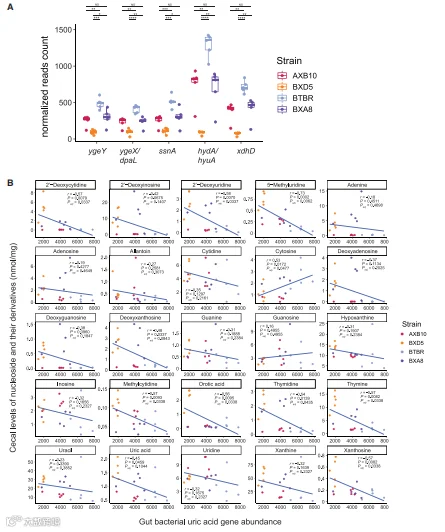
图7. ApoE KO移植小鼠盲肠嘌呤与肠道细菌编码尿酸降解基因的相关性
讨 论
在这项研究中,我们试图识别微生物调节的参与动脉粥样硬化进展的代谢物。我们最初的无菌小鼠移植研究显示,包括UA在内的几种嘌呤的水平受到肠道微生物的影响,并与动脉粥样硬化负担相关。这些初步结果促使我们更深入地探索肠道菌群对嘌呤稳态的作用,并使我们能够识别能够厌氧分解嘌呤的细菌,并发现一簇细菌基因,这些基因对这些底物的厌氧分解是必需的。我们还证明了编码这些功能的类群降低了小鼠的循环UA水平。
总之,这项工作不仅对动脉粥样硬化有意义:它提供了肠道细菌代谢如何影响循环中UA的见解,并表明能够厌氧分解嘌呤的微生物是宿主嘌呤稳态的重要驱动因素,无论是局部(即肠道)还是全身性的。一项最近的预印本使用互补的方法,包括高尿酸血的人类和梭菌属物种的突变,确定了与UA降解相关的相同的细菌基因簇,并得出了类似的结论,关于它对宿主UA稳态的作用。
在人类中,近三分之二的嘌呤是内源性产生的,而剩余的部分来自饮食,主要被十二指肠吸收,但由于核苷的转运体在上皮细胞中表达,在大肠中也可能发生吸收,而且当嘌呤的需求增加时,它们的丰度也会增加。例如,在结肠癌中,被吸收的嘌呤可以被肠细胞或结肠细胞利用或降解为UA。在大多数哺乳动物中,UA可以进一步代谢为尿囊素,但在类人猿中,存在一个带有多个突变和早期终止密码子的尿酸酶基因,导致UA的积累。由于UA相对不溶,人类容易患上由UA沉淀引起的疾病,包括痛风和肾结石。
事实上,美国成年人中高尿酸血的患病率约为20%,并且在最近几十年中一直在稳步增加。这种增加可能与高嘌呤饮食、果糖饮料(已知会增加UA水平)和酒精消费的普遍性有关。
细菌的厌氧嘌呤利用在100多年前就被首次描述,但相对较少的物种被鉴定,而且这些分离物是从环境来源获得的,是必需的嘌呤分解的厚壁菌。在遗传操作出现之前,就已经确定了它们的嘌呤分解途径的生化学,而且只有在过去的十年里,它们的基因组才被测序,尽管编码这一过程的遗传元件仍未明确。更多营养多样的厌氧嘌呤利用生物,包括从白蚁肠道分离出来的厚壁菌和变形菌,代谢嘌呤,并显然在氮限制的宿主饮食中回收嘌呤氮,但这些生物的进一步生化和遗传分析尚未发表。
通过体外分析,通过单个基因的过表达,或者通过使用含有额外的碳和能量来源的复杂培养基培养的细胞悬液和培养物,鉴定了编码厌氧嘌呤降解的拟议途径的操纵子。虽然这些努力确定了编码厌氧嘌呤分解所必需的功能的适当的基因组区域,但没有制定出允许在多种嘌呤上可重复生长的适当条件,而且对编码的代谢的完全理解仍然难以捉摸。
我们研究了UA在多大程度上可以作为肠道细菌的碳和能量来源,以及肠道菌群组成可能在多大程度上影响宿主的全身嘌呤浓度。我们测试了来自6个门的34个人类肠道分离物,认识到(1)微量元素(Mo和se)对代谢功能的要求,和众所周知的分解代谢物抑制现象,据报道大肠杆菌的yge操纵子也可能存在于其他细菌中。
我们确定了三个不同门的代表,主要属于厚壁菌和变形菌,它们能够轻易地降解UA。有一小部分生物被证明可以利用腺嘌呤和次黄嘌呤,并且,独立于使用UA或腺嘌呤的能力,包括E. coli在内的一些生物在提供尿囊素的培养基中厌氧生长(图4,S4和S5A),与描述该化合物只作为氮源的报告相反。然而,这些特性并不一致地存在于任何一个分类群中,甚至在物种之间也发现了菌株差异。
此外,另一种碳源(葡萄糖和/或果糖)的存在降低或消除了UA的代谢,证实了碳水化合物降解抑制的转录调节,并表明了一种可能调节肠道中嘌呤利用的营养参数。
在两种变形菌中,利用UA的能力受到Se和Mo的存在的影响(图S5B)。然而,在C. difficile和E. bolteae中,金属的效应并不明显,可能反映了E. bolteae的RNA-seq数据中鉴定的多种金属摄取系统的高表达(图S7),以及影响其他嘌呤分解的厚壁菌中矿物质限制的多种金属限制转移的要求。
有人认为,膳食Se可能影响人类肠道菌群对嘌呤的代谢,但这里显示的体外结果表明,任何需求可能取决于菌群组成:如果嘌呤利用者主要是变形菌的成员,那么是必需的,但如果厚壁菌占优势,那么可能不太重要。
总之,这些结果(1)表明,提示系统发育不能很好地预测微生物嘌呤的利用;(2)表明,鉴定的基因的存在与生物利用的嘌呤的广度没有相关性;(3)证明了两种营养参数,即碳源和金属可利用性,对嘌呤代谢的影响;(4)强调在预测肠道微生物群的嘌呤代谢时,需要进行基因组学之外的评估。
最后,需要注意的是,虽然使用能够降解嘌呤的细菌可能是降低促炎UA的一个吸引人的策略,但还需要更多的工作来完全理解对宿主的影响。例如,肠道上皮是成年哺乳动物最活跃的自我更新组织,对核苷酸的需求很高,因为它们是增殖和能量所必需的。腺嘌呤——我们研究中的几个分类群消耗的一种嘌呤(图4和S4)——是无法从头合成嘌呤的肠道细胞中核酸的前体。
此外,Colgan小组的最近的工作表明,肠道细菌是肠道粘膜用于核苷酸生成的嘌呤的主要来源。重要的是,通过细菌定殖直接补充嘌呤,改善了肠道上皮细胞的创伤愈合和屏障恢复能力,并表明嘌呤对结肠上皮增殖、能量平衡和粘液屏障完整性起着重要作用。腺嘌呤还抑制了肠道上皮细胞中的TNF(组织坏死因子)-α信号传导,并减少了在葡聚糖-硫酸钠诱导的结肠炎小鼠模型中的粘膜炎症。与这些结果一致,一项最近的研究发现了与肠易激综合征相关的肠道嘌呤饥饿。
我们的结果显示,PDB可以代谢多种嘌呤,并降低肠道中次黄嘌呤的水平,以及编码厌氧嘌呤降解中关键蛋白的基因的丰度与肠道中嘌呤的降低相关,表明这些生物对肠道上皮细胞和屏障功能的嘌呤可利用性的影响需要仔细检查,特别是考虑到许多被鉴定为厌氧嘌呤降解者的分类群,包括E. coli、E. bolteae、F. varium和C. difficile,已经与疾病相关。因此,有必要进一步研究嘌呤降解对这些分类群的适应性和宿主健康的贡献。
总之,这里展示的工作表明,厌氧嘌呤利用在肠道居住的细菌中是普遍存在的,并表明微生物嘌呤降解者是宿主肠道和循环中UA水平的嘌呤稳态的重要调节因素。需要进行研究,以剖析有氧与厌氧嘌呤消耗途径对嘌呤经济、肠道生态和包括动脉粥样硬化在内的健康状况的贡献。
本文译自:
Kasahara K, Kerby RL, Zhang Q et al. Gut bacterial metabolism contributes to host global purine homeostasis. Cell Host Microbe. 2023, 31(6):1038-1053.

往期推荐
2. 奇辉生物科技-企业介绍

声明:原创不易,转载请注明出处,谢谢合作。