
奇辉观点
肠道微生物特征与2型糖尿病(T2D)之间的关联一直不一致,这部分是因为这种疾病的复杂性以及研究设计的差异。即使在某些情况下,特定的微生物种类与T2D有关,但基于特定微生物菌株的机制也无法归因于这些关联。本研究进行了一项全面的T2D微生物组研究,分析了来自美国、欧洲、以色列和中国10个队列的8117个个体的随机宏基因组,发现在19个系统发育多样化的物种中,T2D与它们的失调有关,如富集的梭菌属(Clostridium bolteae)和减少的丁酸弧菌(Butyrivibrio crossotus)。这些微生物还促成了可能潜在T2D发病机制的群落层面功能变化,如葡萄糖代谢的扰动。研究还识别了27种物种的菌株内系统发育多样性,这些多样性解释了T2D风险的个体间差异。在某些情况下,这些差异可以通过菌株特异性基因携带来解释,包括涉及水平基因转移的各种机制和代谢风险背后的新生物学过程的位点,如群体感应。此研究以菌株解析的方式提供了稳健的跨队列微生物特征,并为T2D提供了新的机制见解。研究结果不仅有助于理解肠道微生物群与T2D之间的复杂关系,而且可能有助于开发新的诊断工具和治疗方法。通过对肠道微生物群的深入分析,这项研究为未来的临床应用奠定了基础,可能有助于预防和治疗T2D。
论文ID
本文译自:Mei Z, Wang F, Bhosle A, et al. Strain-specific gut microbial signatures in type 2 diabetes identified in a cross-cohort analysis of 8,117 metagenomes. Nat Med. 2024 Jun 25. doi: 10.1038/s41591-024-03067-7.
发表杂志:Nat Med
影响因子:58.1
通讯作者:Dong D. Wang
作者单位:Harvard Medical School
引 言
2型糖尿病(T2D)全球大约影响5.37亿人。它的特征是β细胞质量和功能的逐渐下降,通常伴随着低度系统性炎症和胰岛素抵抗。在过去二十年中,肠道微生物组越来越被认为是一个代谢活跃的“器官”,位于宿主遗传和环境因素的交汇点。尽管人类研究已经识别出T2D的多种微生物特征,但现有的发现大多是不一致的,部分原因是由于研究人群规模小以及不同研究中的设计和分析方法的差异。此外,早期研究未能充分调整T2D的主要风险因素和混杂因素,例如二甲双胍的使用和肥胖,限制了观察到的关联的有效性,同时导致了变异。因此,需要在大型人群中进行标准化数据处理和分析的研究,以阐明肠道微生物及其相应的分子活动如何贡献于T2D的病理学。
微生物群落结构和特定物种以前已经与代谢风险因素以及T2D联系起来,然而,致病机制可能是特定菌株的,这意味着特定的微生物菌株与疾病结果有因果联系,或者导致宿主疾病发展的微生物功能过程是由微生物物种中的一个菌株子集执行的。大肠杆菌(Escherichia coli)是密切相关菌株之间微生物生理学截然不同的经典例子,它包括从无害(例如,K12菌株)到致病(例如,肠出血性大肠杆菌O157:H7)到益生菌(例如,Nissle 1917菌株)的菌株。理解特定菌株的机制对于T2D尤其重要,T2D是一种具有强烈饮食和炎症基础的疾病,因为宿主饮食和免疫系统在塑造肠道微生物物种内变异中是关键的选择压力因素。然而,对于T2D中的亚种水平微生物特征和特定菌株功能进行全面调查以获得深入的机制见解存在高度未满足的需求。
在这里,我们展示了来自我们新成立的微生物组与心血管代谢疾病(MicroCardio)联盟的10个队列的8117个宏基因组的元分析,这些队列包括了来自美国、欧洲、以色列和中国的T2D患者、糖尿病前期和正常血糖状态的个体。我们首先对原始宏基因组测序数据进行了统一的生物信息学重新处理和批次效应校正,并协调了不同队列中糖尿病和糖尿病前期终点的诊断。接下来,我们识别了在病例和对照组之间差异丰富的特定肠道微生物物种和功能(即微生物编码的酶和途径)(假发现率(FDR)<0.10)。最后,为了更深入地了解T2D中物种内系统发育多样性和特定菌株功能基因携带的影响,我们应用了一系列菌株解析分析方法。我们的研究采用了功能聚焦和菌株解析的方法,代表了在种族和地理上多样化的人群中对T2D微生物组的全面调查。
结 果
全球人群的数据协调
我们统一处理了来自美国、以色列、瑞典、芬兰、丹麦、德国、法国和中国的10个队列的序列和表型数据,包括四个从头生成的数据库和六个已发表的数据集(图1a,扩展数据图1和补充表1)。我们最终的数据集包括来自1851名T2D患者、2770名糖尿病前期参与者和2277名正常血糖参与者的8117个宏基因组(两个队列包括重复的微生物组采样,见方法),包括女性和男性(女性,54.4%),年龄范围广泛(平均值±标准差,57.9±10.7岁)和体重指数(BMI,28.6±5.8kg/m²,图1a,扩展数据图1和补充表1)。我们使用美国糖尿病协会的诊断标准协调了病例-对照状态,基于空腹血糖、2小时口服葡萄糖耐量试验、血红蛋白A1C和药物使用(方法),T2D的主要风险因素,例如BMI,以及血液样本中的代谢和炎症实验室测试,例如高灵敏度C反应蛋白(hs-CRP),跨队列协调。使用bioBakery 3.0工作流程,我们基于测序数据生成了分类学和功能谱(生化途径和酶)。在识别的微生物物种中,30.9%存在于所有包含的队列中(“普遍物种”),52.9%存在于2-9个队列中(“重叠物种”),16.2%是特定队列中独有的(“独特物种”),图1b。正如预期的那样,肠道微生物组的大部分变异是由拟杆菌门与厚壁菌门之间的权衡驱动的(图1c)。有关微生物特征的人口分布的更多细节见补充文本。我们应用了MMUPHin工作流程,该工作流程减少了批次效应解释的方差,通过置换多元方差分析(PERMANOVA)量化,从8.4%降低到4.0%,同时保留了生物学上有意义的个体间变异(扩展数据图2)。此外,我们采取了保守的元分析方法,即在每个队列内分别进行分析,并在所有队列中汇总统计数据,在下游分析中进一步调整潜在的批次效应。
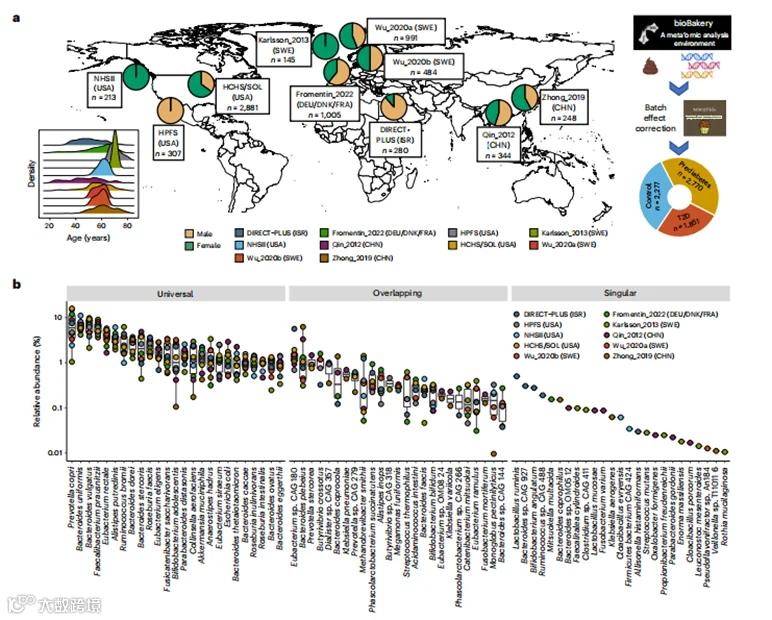
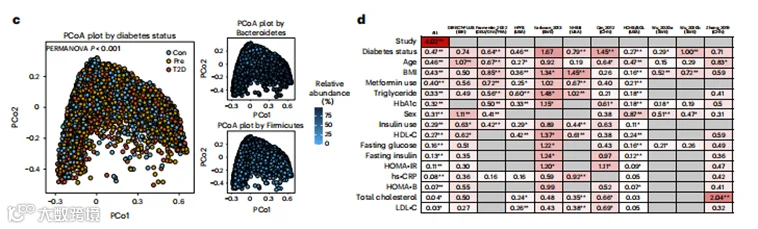
图1 | 与2型糖尿病(T2D)相关的微生物群落结构概述。
a, 为了研究T2D中的肠道微生物组,我们从涵盖八个国家的10个队列中组装了一个宏基因组测序数据集。数据集包括来自MicroCardio联盟新建立的1851名T2D患者、2770名糖尿病前期个体(Pre)和2277名正常血糖对照组(Con)的8117个宏基因组。研究人群包括女性和男性(女性占54.4%),年龄范围广泛(平均57.9岁)和BMI(平均28.6 kg/m²),以及不同的种族/族群,如亚洲人、白人和拉丁美洲出生的美国拉丁裔移民。我们应用了bioBakery 3.0工作流程21来处理测序数据,进行统一的分类学和功能分析,并使用MMUPHin框架22来校正批次效应。此图示由BioRender.com创建。
b, 展示了前25个普遍存在的(在所有10个队列中都存在的)、重叠的(至少在两个队列中存在的)和独特的(仅在一个队列中发现的)物种在每个队列中的相对丰度均值。箱线图的中心显示中位数,箱体表示其四分位间距(IQR),上下限须分别表示上四分位数上方1.5×IQR和下四分位数下方1.5×IQR。
c, 主坐标分析(PCoA)显示了微生物群落配置与T2D之间显著的关联,以及预期的拟杆菌门和厚壁菌门之间的权衡,其中PCo1轴解释了所有三个图中18.7%的变异,PCo2轴解释了8.4%的变异。PCoA基于物种水平的Bray-Curtis差异性。d, 通过PERMANOVA(999次排列)量化研究效应、T2D状态、协变量和循环生物标志物在分类学上解释的变异比例,基于物种水平的Bray-Curtis差异性。在三项研究中(Wu_2020a10、Wu_2020b10和Zhong_2019(参考文献11))没有关于二甲双胍使用的数据,因为它们只招募了新诊断的、未经治疗的T2D和糖尿病前期的参与者。所有统计测试均为双尾。*P<0.05,**P<0.01。HDL-C,高密度脂蛋白胆固醇;HOMA-B,β细胞功能稳态模型评估;HOMA-IR,胰岛素抵抗稳态模型评估;LDL-C,低密度脂蛋白胆固醇。
微生物配置、物种与2型糖尿病
我们首先评估了整体微生物组配置与2型糖尿病状态之间的关联。尽管病例-对照状态不是微生物组整体结构变异的主要驱动因素(图1c),但PERMANOVA表明它在分类学方面(解释方差的百分比(R2) = 0.47%;P < 0.001)、生化途径(R2 = 0.47%;P < 0.001)和酶谱(R2 = 0.30%;P < 0.001)方面的关联是显著的。此外,在2型糖尿病状态、协变量和循环生物标志物中,2型糖尿病状态占微生物组组成变异的最大比例(图1d;详细的PERMANOVA结果见补充文本)。
为了识别协调的物种级标志,我们使用MaAsLin2中的回归模型(参考文献23)来识别在每个队列中T2D状态不同分布的微生物特征,并使用元分析汇总了队列间模型的效应估计(方法)。我们的主要模型将病例-对照状态按T2D、糖尿病前期或正常血糖对照顺序分类(扩展数据图3a)。其次,我们将病例-对照状态作为二元变量(T2D或正常血糖对照)在排除糖尿病前期个体的亚人群中建模(扩展数据图3b)。我们的元分析鉴定了19个系统发育多样化的物种与T2D显著相关(FDR<0.10),独立于年龄、性别、BMI和二甲双胍使用,经过多重假设检验校正(我们称之为“生物标志物物种”,图2a,b)。在这19个生物标志物物种中,有5个与T2D相关,14个与糖尿病前期和T2D都相关。我们在补充表2和3中包含了所有分析的微生物物种的元分析和队列特定结果。值得注意的是,与先前的T2D宏基因组研究相比,我们的研究新鉴定了14个物种,而5个物种之前已有报道,包括3个T2D富集物种,柠檬酸梭菌(Clostridium citroniae)、梭菌属(Clostridium bolteae)和大肠杆菌(Escherichia coli),以及2个T2D减少物种,Coprococcus eutactus和Turicibacter sanguinis。大多数生物标志物物种在正常血糖对照、糖尿病前期个体和T2D患者中的丰度呈现出一致的上升或下降趋势(图2c),这由它们的序数模型中的显著斜率表明(图2a)。
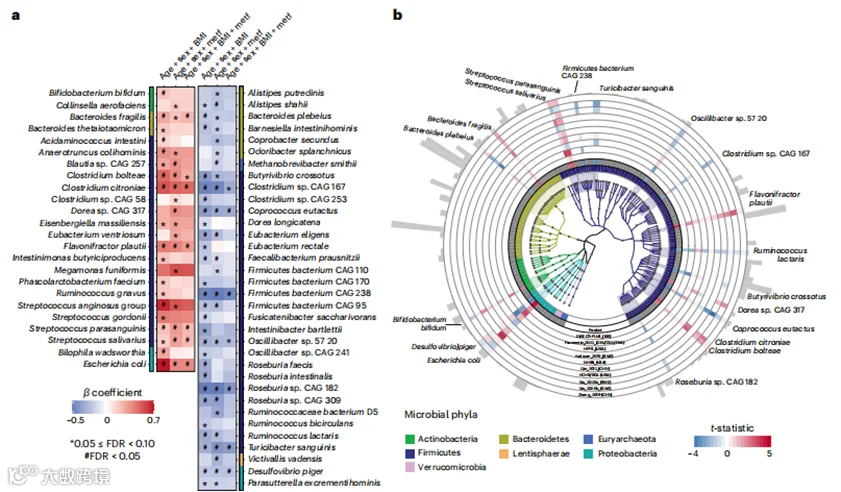
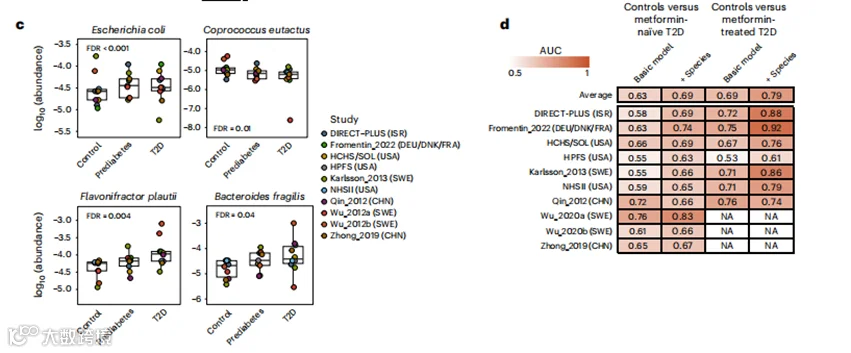
图2 | T2D的跨队列微生物特征。
a, 基于来自1851名T2D患者、2770名糖尿病前期个体和2277名正常血糖对照组的8117个宏基因组的微生物物种与T2D的元分析关联。蓝色至红色的渐变代表了通过包含疾病状态作为序数变量(正常血糖对照组、糖尿病前期或T2D)的线性混合模型量化的关联的强度和方向,并调整了年龄、性别、BMI和二甲双胍(metf)使用。为了多重比较校正,我们以0.10的目标率控制了FDR;*0.05 ≤ FDR < 0.10; #FDR < 0.05。
b, 与T2D显著相关的系统发育多样性微生物物种。蓝色至红色的渐变代表了微生物物种与T2D表型的关联。最内圈的颜色和系统发育树区分了主要的门类。最外圈条形的高度与微生物物种的相对丰度均值成比例。我们展示了调整了前述协变量的序数和二元模型的显著结果(FDR < 0.10,方法)。
c, 微生物物种与T2D状态之间的选择性剂量-反应关联。箱线图的中心显示了队列特定相对丰度的中位数,箱体表示它们的四分位间距(IQR),上下限须分别表示上四分位数上方1.5×IQR和下四分位数下方1.5×IQR。统计模型、多重比较校正的方法和样本大小与a中的相同。a、b和c中的所有统计测试均为双尾。
d, 微生物物种的纳入提高了随机森林模型在分类二甲双胍治疗或未经治疗的T2D与对照组中的性能。AUC是通过应用在该行所有队列之外训练的模型并在该行的队列中验证获得的。基础模型包括年龄、性别和BMI,而其他模型还包括微生物物种。在二甲双胍使用者中,Wu_2020a、Wu_2020b和Zhong_2019的AUC不可用(NA),因为他们只招募了未经治疗的参与者。
我们鉴定了两种链球菌属(Streptococcus spp.),它们是口腔的常见居民,在T2D个体中富集,这表明口腔微生物可能转移到肠道,这指示了一种促炎状态。此外,我们发现产生免疫原性脆弱菌素(fragilysin)的拟杆菌属(Bacteroides)脆弱菌种(B. fragilis)在T2D病例中富集。相比之下,另一种拟杆菌属物种,拟杆菌属(Bacteroides)普通菌种(B. plebeius),主要在非白人T2D患者中减少。这与最近对B. plebeius与其他拟杆菌属(Bacteroides spp.)表型差异的确认以及其随后被重新归类到海豹杆菌属(Phocaeicola)一致。其他T2D减少的物种包括产生丁酸的细菌和植物衍生多糖的主要降解者,如振荡菌属(Oscillibacter)sp. 57_20、Coprococcus eutactus、乳果糖菌(Ruminococcus lactaris)和丁酸弧菌(Butyrivibrio crossotus)。我们在正常血糖对照组中发现T. sanguinis的丰度增加,这证实了之前关于它通过改变宿主胆汁酸和脂质代谢与低代谢风险相关联的报告。
接下来,我们确定了包含微生物物种在多大程度上使用随机森林模型改进了T2D状态的分类,并使用留一数据集出的方法测试了模型。在随机森林模型中,将未使用二甲双胍的T2D与正常血糖对照组进行分类,我们发现从只包括年龄、性别和BMI的基础模型(平均AUC,0.63)到另外包括微生物物种的模型(平均AUC,0.69,图2d)在接收者操作特征曲线下面积(AUC)上有中等程度的提高。将接受二甲双胍治疗的T2D与对照组进行分类的模型在基础模型中平均AUC为0.69,在包括微生物物种的模型中平均AUC为0.79。
我们确认了在这一国际人群中,二甲双胍的使用强烈混淆了微生物与T2D之间的关联,而进一步调整胰岛素使用和BMI只是略微改变了这些关联的强度(扩展数据图3)。我们发现接受二甲双胍治疗与未接受二甲双胍治疗的T2D患者之间存在不同的微生物组成(PERMANOVA P<0.001,扩展数据图4a),并且在进一步调整二甲双胍使用后,许多微生物物种的关联性减弱(图2a)。在随后的分析中,我们鉴定了10个二甲双胍的微生物特征,即那些仅与T2D病例中二甲双胍使用相关的(扩展数据图4b)。我们通过证明初步分析中物种-T2D关联的beta系数与从排除接受二甲双胍治疗的T2D患者的敏感性分析中计算出的beta系数之间高度相关(斯皮尔曼相关系数,0.95;扩展数据图4c),确认了我们的建模方法有效地解决了二甲双胍使用带来的混淆。
为了解决已识别的微生物特征可能部分捕捉到长期糖尿病状态及其并发症的担忧,我们进行了两项敏感性分析,包括利用拉丁裔社区健康研究/拉丁裔研究(HCHS/SOL)中的新发T2D病例进行的前瞻性分析,以及对未使用胰岛素的T2D病例的分析。这两项敏感性分析得出的关联在很大程度上与主要分析的结果一致(扩展数据图5;敏感性分析的详细信息见补充文本)。此外,我们观察到微生物特征的丰度在正常血糖对照组、糖尿病前期和T2D中的显著剂量-反应效应(图2a),为T2D的微生物生物标志物提供了进一步的证据。总体而言,这些分析表明,已识别的微生物特征不太可能反映T2D的长期持续时间或其并发症。
我们的其他主要发现包括在T2D患者中上调的免疫调节细菌结构成分的生物合成。我们发现,在T2D患者中,两个位于生产高度促炎性脂多糖(LPS)和壁酸的上游途径,即脂质IVA生物合成(NAGLIPASYN-PWY)和聚(甘油磷酸)壁酸生物合成(TEICHOICACID-PWY),是富集的。这也适用于参与LPS和壁酸生物合成的单个酶,如肽聚糖糖基转移酶(EC 2.4.1.129)和脂质IVA 4-氨基-4-去氧-L-阿拉伯糖基转移酶(EC 2.4.2.43,图3b)。参与LPS前体生物合成的酶的丰度与循环高敏C反应蛋白(hs-CRP)水平之间的正相关性,进一步支持了它们的免疫原性(扩展数据图6a和6b以及补充表7)。支持之前报告称拟杆菌的膜成分特别免疫原性,发现一群多样化的拟杆菌属(Bacteroides spp.)编码这些途径和酶。
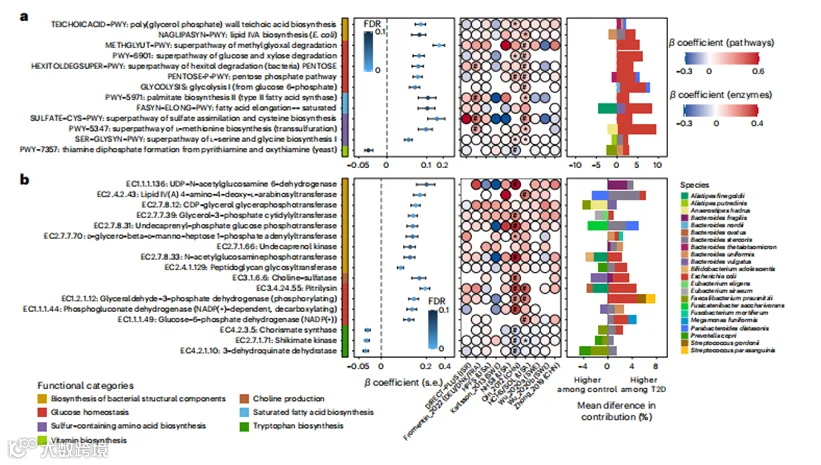
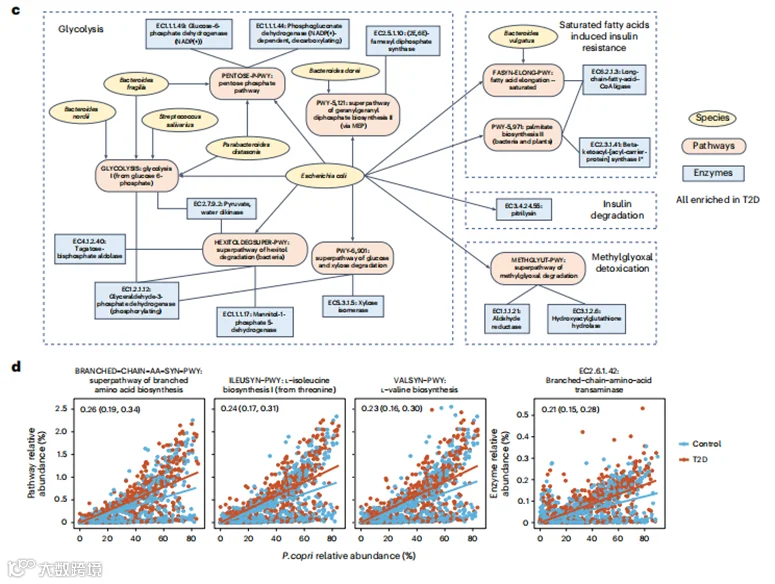
图3 | 参与T2D发病机制的多样化微生物过程。
a, 基于元分析的微生物功能(作为MetaCyc途径)与T2D的关联。
b, 基于元分析的酶(作为EC编号)与T2D的关联,这些功能和酶涉及葡萄糖稳态、硫代谢以及细菌结构成分、B族维生素和必需氨基酸的生物合成。分析中包括了来自1851名T2D患者、2770名糖尿病前期个体和2277名正常血糖对照组的8117个宏基因组。Beta系数来源于多变量调整的线性混合模型(方法),该模型将T2D状态作为自变量,将微生物途径或酶的丰度作为因变量。缺失的圆圈表示在该队列中未检测到的特征。所有结果均通过控制Benjamini-Hochberg方法的FDR,以0.10为目标率进行了多重假设检验校正。*0.05 ≤ FDR < 0.10; #FDR < 0.05。s.e., 标准误差。
c, 显示T2D中胰岛素抵抗、糖酵解和葡萄糖摄取之间相互关系的微生物特征网络。该网络包括与T2D显著相关(FDR < 0.10)的精选MetaCyc途径和酶,以及编码这些微生物功能的物种。
d, T2D患者中的Prevotella copri菌株更有可能携带支链氨基酸生物合成的途径和酶。蓝色和红色线条分别代表对照组参与者和T2D参与者中相对丰度的P. copri与由P. copri编码的特定途径或酶的相对丰度之间的关联,这些线条通过线性回归拟合。左上角的数值是案例-对照状态之间途径丰度对数转换后的差异的后验差异值和98%后验差异区间,由anpan中的混合效应模型确定(方法)。该模型使我们能够在控制其物种水平丰度的同时,识别由P. copri编码的在T2D患者和对照组之间差异丰度的微生物功能。
普雷沃菌属(Prevotella copri)亚群与2型糖尿病
普雷沃菌属(Prevotella copri)已被发现与代谢风险有关,并且曾有假设认为P. copri致病潜力是亚群依赖性的,并且特定于人群。因此,我们试图确定P. copri与T2D之间的关联是否因其具有不同功能潜力的亚群而有所变化。我们首先在anpan(微生物系统发育和基因分析,见方法)中应用了针对特定途径的随机效应模型,以识别P. copri编码的在T2D患者与对照组之间差异丰富的功能,同时控制其物种水平的丰度。我们发现,T2D患者中的P. copri更有可能携带支链氨基酸(BCAAs)的生物合成功能,这是一组致糖尿病的代谢。这些功能包括支链氨基酸生物合成的超途径(BRANCHED-CHAIN-AA-SYN-PWY)、L-缬氨酸生物合成(VALSYN-PWY)、L-异亮氨酸生物合成I(ILEUSYN-PWY,图3d),以及一个执行支链氨基酸生物合成最后一步的多功能酶(EC 2.6.1.42:支链氨基酸转氨酶)。
由于已知P. copri具有离散的亚种结构,我们根据已发布的参考泛基因组(方法,扩展数据图7a)对它的四个亚群进行了分析。正如预期的那样,在欧洲和美国的非西班牙裔白人参与者中,P. copri以A群为主,而在中国人、以色列人和美国西班牙裔人群中主要观察到所有亚群的共存(扩展数据图7a)。然后,我们测试了BCAA生物合成功能在不同亚群中的携带情况是否有所不同,发现在以A群为主的P. copri中,这些功能的丰度显著低于所有亚群共存的P. copri(扩展数据图7b)。然而,我们发现T2D患者中未受调控的BCAA生物合成是P. copri A群所特有的,并且在其他P. copri亚群中未被检测到(扩展数据图7c)。综合来看,这表明P. copri的BCAA生物合成能力是亚群和人群依赖性的,这在以分类学为重点或社区层面的微生物功能分析中否则将无法检测到。
物种内系统发育差异与2型糖尿病
接下来,我们使用anpan的系统发育广义线性混合模型(PGLMMs,方法)来评估物种内系统发育在解释T2D风险个体间异质性方面所占的程度(补充表8)。我们观察到,物种内系统发育差异与27个物种的T2D风险个体间差异有关(图4a)。这些遗传结构中的一些与先前的分析一致,而其他的是首次被鉴定出来。正如预期,许多物种包含特定于宿主地理起源的亚种(扩展数据图8),这与“距离隔离”假说一致,该假说提出宿主和微生物的共同扩散有助于物种的遗传分层。由于这一点,为了确保物种内系统发育结构与T2D风险之间的关联不受种族地理学的混淆,我们在随后的模型中包括了队列成员身份作为协变量。
在直肠真杆菌(E.rectale)中,我们观察到来自中国南部的菌株与T2D在亚群B中有很强的关联,该亚群主要由来自中国和美国白人参与者的菌株组成。包含来自不同地理起源菌株的亚群C,在来自美国西班牙裔人群的菌株和另一个来自北欧人群的菌株与T2D之间显示出强关联(图4b)。Coprococcus comes的亚种结构尚未得到很好的研究。我们的系统发育分析将C. comes菌株分为四个离散的亚种亚群。在其由来自中国、以色列和瑞典的菌株组成的亚群A中,来自瑞典和中国东部的两个菌株与T2D有显著关联(图4b)。尽管许多被发现与T2D有很强的关联的菌株簇可以归因于特定的种族和/或地理群体,我们也发现了例外。例如,一个在所有参与队列中都能检测到的Blautia wexlerae菌株,以及另一个来自美国西班牙裔、以色列和瑞典人群的菌株,与T2D有很强的关联(扩展数据图8)。这些发现提供了证据,表明微生物诱导的T2D风险既可以来自特定于人群的微生物遗传学,也可以来自与人群无关的微生物遗传学。显示出强亚群效应的物种中没有一个是T2D生物标志物物种,这表明仅在物种水平上的分析将忽视了关键的微生物-T2D关联。
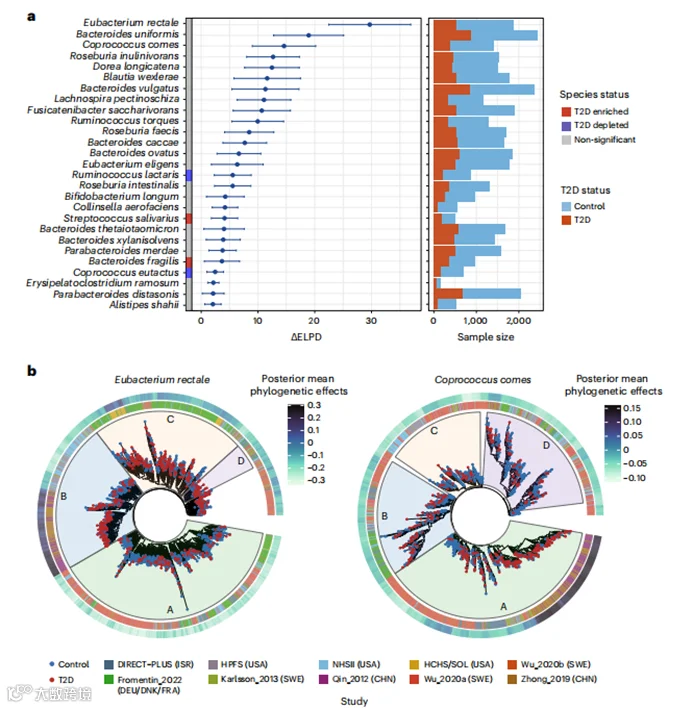
图4:种内系统发育差异解释了微生物物种与T2D风险之间的亚群体特异性和个体化关联。
a, 在27个物种中的每一个中,anpan中的PGLMM(方法)识别了与T2D关联程度不同的亚支。左侧:种内系统发育与T2D之间关联的总结。误差条的中心表示包含和不包含种内系统发育作为随机效应的两个广义线性混合模型之间预期的对数点预测密度(ΔELPD)的差异(方法),误差条表示ΔELPD的标准误差。图中包括的物种是那些ΔELPD大于2的物种。两个模型都调整了年龄、性别、BMI、二甲双胍使用和队列成员身份作为固定效应。anpan通过基于来自蛋白质序列聚类集(UniRef90基因家族)的欧几里得相似性矩阵随机分割边缘来生成种内系统发育树。右侧:经过自适应筛选以去除所关注物种缺失或测序覆盖不足的宏基因组后的T2D患者和正常血糖对照组的样本大小(补充表8和方法)。
b, 两个选定物种的系统发育树,显示了每个物种内亚支与T2D之间的不同关联。内圈表示每个宏基因组的队列成员身份。外圈展示了由PGLMM估计的每个系统发育树叶(宏基因组)的系统发育效应的后验均值,颜色越深表示亚支对T2D风险影响的可能性越高(方法)。A-D,用不同颜色的扇区表示的不同亚支。在anpan中经过自适应筛选后,这种分析的样本大小因物种而异(方法),详细信息见补充表8。
特定菌株的功能变异与2型糖尿病
为了补充物种内系统发育分析,我们利用UniRef90数据,使用anpan中的基因关联模型(补充表9和方法)来识别每个物种中与T2D相关联的基因家族。值得注意的是,在所有在病例和对照之间基因分布不同的物种中,只有大肠杆菌(E. coli)是生物标志物物种;所有其他物种都不是生物标志物物种(图5a)。这表明,如果只关注物种水平的分类特征和社区层面的功能,可能忽略了关键的特定菌株功能。
以大肠杆菌为例,我们检测到的几个菌株簇包含该物种已知亚型的遗传标记(图5b)。值得注意的是,我们在T2D富集的簇中鉴定出编码特定致病性大肠杆菌亚型的毒力因子的基因家族,包括粘附素、侵袭素和毒素。T2D富集的区块5(图5b),包括来自中国和瑞典人群的菌株,富集了涉及各种水平基因转移(HGT)机制的基因家族,如移动遗传元素、噬菌体、共轭和遗传重组,暗示大肠杆菌菌株可能通过HGT获得有利特征和毒力因子。具体来说,鉴定出两个与毒力相关的基因家族:一个与聚集性大肠杆菌(EAEC)中的质粒毒力(UniRef90_A0A376NZ25)相关,EAEC以其诱导炎症反应和粘膜毒性而闻名,另一个包含编码与粘膜和外周水平增强炎症反应相关的LPS生物合成蛋白(UniRef90_A0A3E1VFS6)的基因。在T2D富集的区块3中,我们观察到富集的基因家族包含编码重排热点元件(rhsA;UniRef90_A0A377DDJ9)、它们相关的YD肽重复(UniRef90_A0A376MP41)和vgrG(缬氨酸-甘氨酸重复G)(UniRef90_A0A377LHH4)蛋白,以及Ail/Lom家族蛋白(UniRef90_A0A2G9A1M3)。rhsA编码在肠出血性大肠杆菌(EHEC)[53]中通过依赖vgrG的过程传递到目标真核猎物细胞的免疫蛋白,而Ail/Lom家族蛋白在EAEC中起毒力作用。区块1和4,主要来自中国和美国西班牙裔人群的T2D患者,包含编码特定致病性大肠杆菌粘附因子的基因家族,包括一个假定的粘附素(UniRef90_A0A2Y0X8H8)和一个菌毛蛋白(UniRef90_A0A1Y4J7A6)。唯一的T2D减少的簇(区块2)来自不同的地理起源,包含与微生物对环境压力反应相关的各种基因家族。这些基因可能为菌株提供适应优势,例如逃避有害环境和优化营养利用。
为了扩展这种跨物种的特定菌株生物过程的特征,我们根据基因关联模型结果进行了基因集富集分析(图5a,扩展数据图9和补充表10;方法)。共有31个基因本体(GO)术语与T2D相关(FDR<0.10)。扩展我们在大肠杆菌的发现,与HGT相关的GO术语解释了多个物种中菌株水平的多样性。在T2D患者中,富集的GO术语包括与糖酵解、细菌结构成分的生物合成以及在不利条件下生存至关重要的生物过程有关的生物过程,以及毒力因子和抗生素抗性基因。此外,以直肠真杆菌(E.rectale)为例,我们发现参与鞭毛依赖性细胞运动和趋化的基因在T2D患者中更为普遍,表明特定菌株适应肠道中的氧化应激和炎症,而群体感应的GO术语在正常血糖对照组中富集,表明菌株的生存优势。
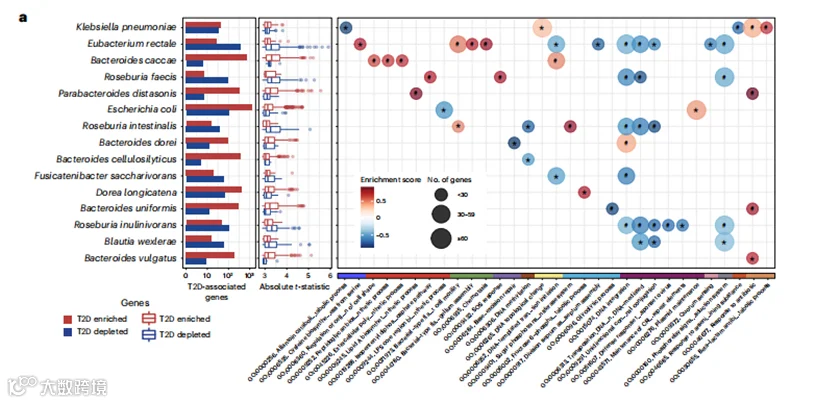
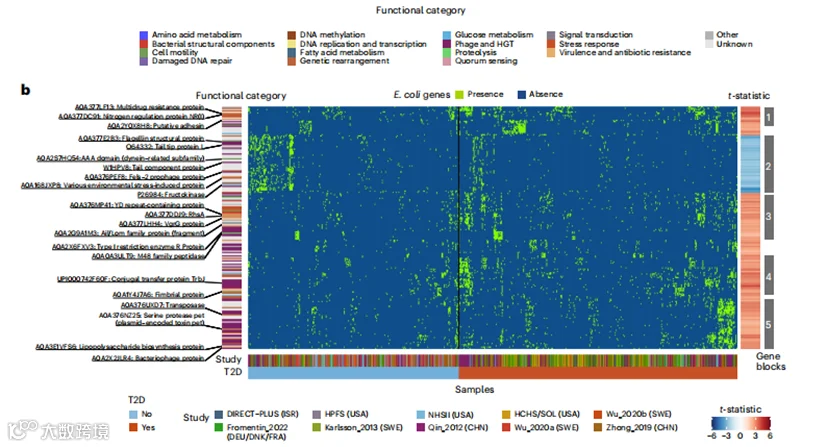
图5 | 菌株特异性基因携带和生物化学作用对T2D发病机制的贡献。
a, 许多差异分布的UniRef90基因家族(蛋白质序列的聚类集)为亚种与T2D不同关联的功能解释提供了依据。条形图显示了经过自适应筛选后与T2D显著相关的UniRef90基因家族的数量。箱线图展示了与T2D正相关(红色)和负相关(蓝色)的UniRef90基因家族效应大小(t统计量)的分布。箱体中心显示了t统计量的中位数,箱体表示它们的四分位间距(IQR),上下限须分别表示上四分位数上方1.5×IQR和下四分位数下方1.5×IQR。右侧面板显示了基于GO术语和anpan中基因关联模型的t统计量,使用1000次排列的基因集富集分析的结果。气泡图展示了GO术语的富集得分和大小。具有正标准化富集得分(NES)的GO术语包含了在T2D患者中上调的UniRef90基因家族。具有负NES的GO术语包含了在T2D患者中下调的UniRef90基因家族。所有结果都通过控制Benjamini-Hochberg方法的FDR,以0.10为目标率进行了多重假设检验校正。*0.05 ≤ FDR < 0.10; #FDR < 0.05。
b. UniRef90基因家族概况显示了E. coli的宏基因组检测菌株。热图显示了与T2D显著相关的基因,每一列代表一个宏基因组,每一行代表一个UniRef90基因家族。颜色表示UniRef90基因家族在宏基因组中的存在(绿色)或缺失(蓝色)。右侧的热图展示了来自anpan中基因关联模型的UniRef90基因家族与T2D关联的t统计量。红色表示在T2D中富集的基因家族,而蓝色表示在T2D中耗尽的基因家族。在anpan中经过自适应筛选后,基因关联模型中的样本大小因物种而异(方法),详细信息见补充表8。
讨 论
在过去十年中,关于肠道微生物组和2型糖尿病(T2D)的大量证据已经出现,这在理解、预防和治疗T2D方面激发了对微生物组潜在临床应用的显著兴趣。然而,文献一直存在不一致性,通常侧重于高水平的群落组成,可能阻碍了更深层次的功能见解和转化努力。为了克服这些挑战,我们进行了一项大型且人口统计学多样化的研究,研究了特征明确的人类群体。我们的研究在MicroCardio联盟中,对基于人群的微生物组研究集合进行了整体微生物群落结构、特定微生物分类和功能特征以及特定菌株功能的评估,这些研究涉及T2D、糖尿病前期和正常血糖状态的参与者。我们的跨队列元分析鉴定了T2D的系统发育多样化的物种水平微生物特征,其中绝大多数是新鉴定的。此外,观察到包括多种途径的社区层面功能变化,如免疫原性细菌结构成分的生物合成上调、糖酵解的扰动,以及丁酸发酵的下调(见补充文本以获取更深入的讨论)。
重要的是,我们的菌株解析分析显示了微生物-T2D关联中的物种内异质性,并鉴定了解释这种异质性的特定菌株功能,例如与水平基因转移(HGT)、支链氨基酸(BCAA)生物合成以及在氧化应激和炎症环境下赋予适应优势的功能有关的那些。尽管动物模型为特定菌株功能在代谢状况中的因果作用提供了多条证据,但以前的T2D微生物组研究没有检查菌株水平的多样性。这主要是由于以前缺乏可靠的菌株鉴定方法和利用它们的统计方法。我们的研究鉴定了T2D患者中一个物种携带的特定基因与其正常血糖对照组的差异。许多已鉴定的功能元素,如与噬菌体、HGT和移动遗传元素相关的那些,属于导致物种内变异的过程,即突变和基因流动,这与关于将遗传变异引入微生物无性繁殖细胞系的已建立知识一致。此外,通过HGT,微生物可以获取有助于肠道微生物进化的新特征,赋予新的表型,如毒力、共生和竞争适应性,这可能导致与T2D有不同关联的菌株。
我们的研究有几个优点,包括大型和多样化的研究人群以及统一的处理和分析方法,以及微生物组菌株流行病学的方法。然而,它的本质上是观察性的,这是许多此类微生物组研究共有的局限性。尽管我们在统计模型中调整了主要混杂因素,但我们无法控制饮食、体力活动、吸烟或除二甲双胍和胰岛素外的其他药物等协变量。此外,尽管我们应用了批次校正和元分析以最小化批次效应,但由于跨队列使用了不一致的样本收集、DNA处理和DNA测序方法,我们无法消除它们的影响。另一个局限性是我们没有根据T2D和糖尿病前期的表型和病理异质性进行亚型分析。此外,尽管我们鉴定了与T2D风险相关并解释了菌株水平功能变异的微生物基因,这些基因涉及对环境选择压力的适应性反应和HGT,但我们的研究没有直接测量特定环境中选择压力或HGT的大小。最后,尽管我们的研究包括了多个独立的人群,并展示了表明跨人群可复制性的队列特定结果,但没有额外的复制队列限制了我们测试发现的普遍性的能力。
尽管我们的研究没有建立因果联系,应该被解释为假设生成,但它提供了迄今为止从人群研究角度来看肠道微生物组参与T2D发病机制的最全面证据。这些结果为未来的机制研究奠定了基础。此外,通过研究微生物菌株的遗传组成和特征,我们对微生物的生物学和致病性有了更细致的理解,使我们更接近因果关系。我们的发现为肠道微生物组在T2D发病机制中的潜在功能角色提供了证据,并强调了未来诊断应用中识别分类学和功能生物标志物的重要性。此外,我们对特定菌株功能基因的检查建立在先前的临床前模型机制研究基础上,我们希望它将促进未来旨在精确描述肠道微生物在T2D发展中作用的研究。
往期推荐
