

论文ID
原名:Machine learning approach reveals microbiome, metabolome, and lipidome profiles in type 1 diabetes.
译名:机器学习方法揭示了T1D中的微生物组、代谢组和脂质组特征
发表杂志:J Adv Res
影响因子:10.479
通讯作者:Jianping Weng
发表单位:中国科学技术大学
摘要:
1型糖尿病(T1D)是一种受遗传和环境因素影响的复杂疾病。肠道微生物组、血清代谢组和血清脂质组已被确定为影响T1D病理生理机制的关键环境因素。
我们旨在通过机器学习探索T1D患者的肠道菌群、血清代谢物和血清脂质特征。
我们评估了137名个体,包括38名T1D患者、38名健康对照和61名用于验证的T1D患者,构成了一个横断面队列。我们使用机器学习方法(逻辑回归、支持向量机、高斯朴素贝叶斯和随机森林)对肠道微生物组、血清代谢物和血清脂质谱进行了表征。
使用菌群组成的机器学习方法不能准确诊断T1D(模型准确率=0.7555),而使用代谢物组成的模型准确率为0.9333。基于代谢物组成,3-羟基丁酸和9-氧代-十八碳二烯酸(曲线下面积分别为0.70和0.67,两者在T1D中均增加)是由多种生物信息学方法筛选出的有意义的重叠代谢物。我们在验证组中确认了微生物组、代谢组和脂质组特征的生物学相关性。
通过使用机器学习算法和多组学,我们证明了T1D患者与改变的菌群、代谢物和脂质组特征或功能相关。
引言:
1型糖尿病(T1D)是一种受遗传和环境因素影响的复杂疾病,它伴随着多种并发症,严重威胁患者的生存和生活质量。值得注意的是,肠道微生物组、血清代谢组和血清脂质组已被确定为影响T1D病理生理机制的关键环境因素。最近基于测序的方法揭示了T1D患者的肠道菌群组成与健康对照有所不同,暗示这种改变可能会导致肠道通透性的增加。此外,最近的研究表明,菌群产生的代谢物可能是T1D发病的潜在保护因素或触发因素,而改变的代谢物(如短链脂肪酸)可能会影响T1D的进展。而且,早期的脂质组研究表明,T1D的发生是由脂质代谢紊乱引起的,而进展到胰岛自身免疫或明显T1D的儿童的脂质组特征与健康对照不同。
以前的研究使用了单一的组学策略来研究环境因素对T1D的影响。青少年糖尿病环境决定因素(TEDDY)研究调查了与T1D相关的肠道微生物组功能谱,其特征是与短链脂肪酸发酵和生物合成相关的微生物减少。以前的代谢组和脂质组研究也表明,肠道微生物可以改变肠道代谢物和脂质,加剧T1D患者血清代谢组和脂质组谱的异常。然而,T1D的肠道微生物组、代谢组和脂质组的全面失调分析和特征还缺乏,而这三个组学之间的相互作用与T1D发病机制的关系还需要进一步鉴定。虽然单一的组学数据提供了有价值的信息,但多组学分析有可能全面地探索临床特征、微生物物种和代谢物之间的关联。此外,与传统的生物信息学分析方法相比,机器学习(ML)方法应用了新的策略来分析多样的组学数据。一般来说,机器学习和多组学分析在T1D研究领域的整合相对缺乏。填补这一空白可能会为T1D的发生和进展提供有价值的见解。
在当前的工作中,我们进行了全面的分析,将T1D患者的肠道微生物组、代谢组和脂质组数据集进行了整合,旨在深入了解这些组学的格局和关联,以指导T1D的诊断,特别是治疗。为了支持这些结果,我们进一步设计了基于发现和验证数据训练的ML模型,来研究确定的微生物和血清代谢物/脂质在塑造宿主功能状态方面的相关性,以及它们如何影响T1D的发展和结果。
结 果
1、 研究样本的临床特征
本研究采用了一种横断面研究,包括99名T1D患者和38名健康对照。入组细节在方法部分讨论。根据年龄和性别,38名T1D患者(T1D组,36.8%男性)与38名健康对照(HC组,36.8%男性)一对一配对。
剩余的61名T1D患者(验证组,44.3%男性)用于验证在T1D发现组中发现的ML选择的菌群和代谢组特征。参与者的临床特征在表1中呈现。
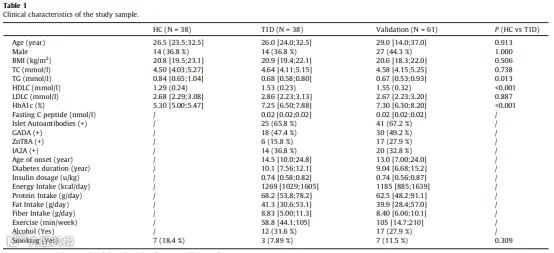
表1 研究样本的临床特征
1、 T1D患者的微生物特征发生了改变
图1A展示了属水平相对丰度的概览。图S1A(置换MANOVA检验,F = 1.1004,R2 = 0.014652,p < 0.324)和图S1B(ACE指数,t检验,p = 0.21685)显示了β-多样性和α-多样性的图,两者都没有显示T1D和HC组之间的统计差异。
我们随后使用LefSe来研究两组之间的微生物组成差异。线性判别分析(LDA)得分(一个用于区分特征的限制)是2。LDA值分布的直方图和不同分类单元的系统发育图分别显示在图1B和C中。T1D组中差异最大的微生物大多数是属于厚壁菌门的梭菌属和芽孢杆菌属的成员。LDA最高的微生物是属于变形菌门的γ变形菌纲的肠杆菌目的成员。
我们还应用了四种ML算法(LR、GNB、SVM和RM),这些算法非常适合有限的样本大小,来筛选差异微生物。为了提高准确性,ML算法在ASV、门、纲、目、科、属和种的水平上进行了执行。LR、GNB、SVM和RM(分别为0.7555、0.6292、0.7319和0.7069)的准确性在属水平上最高。然后,我们应用LR算法来选择前10个候选微生物。候选微生物显示在图1D中。ML选择的前10个微生物特征的ROC分析结果显示在图1E中。
优先选择将ML和LefSe候选生物标志物合并用于筛选严格的微生物。包括Ruminococcus torques、Anaerostipes、Veillonella、Erysipelotrichaceae UCG-003、Blautia和Coprococcus在内的六种重叠微生物的曲线下面积(AUC)分别为0.664、0.659、0.520、0.722、0.679和0.637。此外,Erysipelotrichaceae UCG-003表现出最高的判别值(AUC = 0.722)。
接下来,我们在验证组(图S1C)中验证了ML选择的前10个候选微生物的性能,这些微生物区分了T1D患者和健康人。Anaerostipes、Blautia和Erysipelotrichaceae UCG-003是AUC超过0.60的前10个候选微生物,这意味着这些潜在的微生物有效地区分了T1D和健康人。
此外,糖化血红蛋白(HbA1c)与糖尿病的发生和进展密切相关。在我们的研究中,我们发现HbA1c的水平可能会影响与T1D相关的肠道微生物的丰度。
在这些微生物中,如图1C和图1D所示,HbA1c的水平与Ruminococcus torques(图S3A)、Lactobacillales(图S3B)、Streptococcaceae(图S3C)、Bacilli(图S3D)、Erysipelotrichales(图S3E)和Erysipelatoclostridiaceae(图S3F)的比例呈正相关。
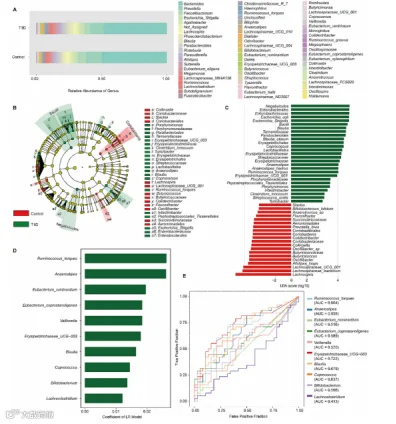
图1 T1D中微生物特征的改变。
(A) 每种微生物在属水平上的丰度的堆叠条形图。
(B) 表示T1D患者和健康人之间有显著差异的微生物类群的系统发育图。
(C) 显著差异的微生物类群的LDA值分布的直方图。
(D) 机器学习(ML)预测的与疾病相关的前10个微生物特征。
(E) ML预测前10个微生物特征的ROC曲线。
3、T1D患者的代谢特征发生了改变
接下来,我们用WGCNA(图2C)确定了高度相关的血清代谢物的模块。WGCNA识别出了六个模块,其中包含了706种血清代谢物的黄色模块与T1D状态最相关。
然后,我们在人类代谢组数据库中对706种血清代谢物进行了注释,以进行通路分析,其中97种代谢物被注释。在对97种代谢物的通路分析中,KEEG数据库中注释了22种代谢通路(图2D)。
我们还使用了ML算法(LR、GNB、SVM和RM)来筛选T1D中改变的代谢特征。LR、GNB、SVM和RM的准确率分别为0.8805、0.8805、0.9333和0.8819。候选代谢物显示在图2E中。对前10个代谢物(图S2A)的ROC分析显示,5-[2-45]-3,4,7,7a-四氢环戊[b]吡喃-6(2 h)-酮(AUC = 0.886)和D-果糖(AUC = 0.882)具有最高的判别值。
接下来,我们在验证组(图S2B)中验证了ML选择的前10个代谢物,其AUC结果与T1D组相似。5-(2-噻吩基)-3,4,7,7a-四氢环戊[b]吡喃-6(2 h)-酮(AUC = 0.833)和D-果糖(AUC = 0.831)在验证组中也表现出最高的判别值。
接下来,我们将T1D中上调的代谢物、ML的前100个候选生物标志物和WGCNA的黄色模块代谢物合并起来。有七种重叠的代谢物,我们对这七种重叠的代谢物进行了ROC分析(图2E)。3-羟基丁酸是AUC最高的代谢物(图2G,AUC = 0.697)。
然后,我们对验证组中的七种重叠代谢物进行了ROC分析(图S2C)。3-羟基丁酸的AUC在验证组中也很高(AUC = 0.780),这意味着它能有效地将T1D与健康状态分开。
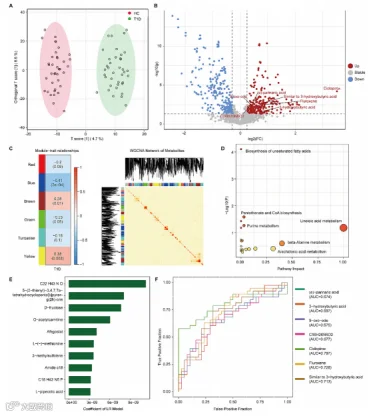
图2 T1D中代谢特征的改变。
(A) OPLS-DA显示了代谢物组成的区分趋势。
(B) 火山图显示了通过倍数变化(>1.2)和p值(<0.05)使用t检验的差异代谢物筛选。
(C) WGCNA的结果。模块-性状关联(左)显示了每个代谢物模块与T1D性状的关系。每个单元格包含相应的相关性和p值。代谢物网络的热图(右)描绘了分析中所有代谢物之间的拓扑重叠矩阵(TOM)。颜色越深表示重叠越高。
(D) 差异代谢物的代谢途径。
(E) 机器学习(ML)预测的对疾病有贡献的前10个代谢特征。
(F) 用三种方法得到的七种重叠代谢物的ROC曲线。
4、T1D患者的脂质组特征发生了改变
根据OPLS-DA(图2A),T1D组和HC组之间的血清脂质组成有所不同。与HC组相比,T1D组有63种差异性血清脂质降低,31种差异性脂质增加(组合倍数变化> 1.2,p值< 0.05)。结果在图3B中显示。
接下来,我们还使用了ML算法(LR、GNB、SVM和RM)来筛选T1D中改变的脂质特征。LR、GNB、SVM和RM的准确率分别为0.8138、0.8138、0.7875和0.7486。
然后,我们使用LR算法来识别10种差异表达最大的脂质。前10种脂质是PC(36:4e)(rep)、TG(16:0/16:0/18:1)、PC(36:4)(rep)、PC(38:6)(rep)、SM(d36:2)、SM(d42:2)、SM(d36:1)、PC(38:6)、SM(d42:1)(rep)和PC(34:1)(rep)(图3C)。
然而,T1D组和验证组中前10种脂质的ROC分析显示,前10种脂质的AUC并不显著。
通过将ML的前100个候选脂质和倍数变化的94个差异脂质合并,筛选出了10种重叠的脂质:TG(18:1/18:1/18:2)、ChE(18:1)、PE(18:1p/18:2)、PE(36:4e)、PC(38:6e)(rep)、PE(34:1e)、PC(42:8)、PI(16:0/18:1)、TG(18:1/18:2/18:2)和PC(39:5)。此外,PE(18:1p/18:2)、PE(36:4e)和PC(39:5)也是根据倍数变化筛选出的前10个差异脂质。
接下来,我们将ML的前100个候选生物标志物和倍数变化大于1.2且p值小于0.05的94种差异性血清脂质合并起来。
我们在人类代谢组数据库中对这些合并的脂质进行了注释,以进行通路分析(图3D)。
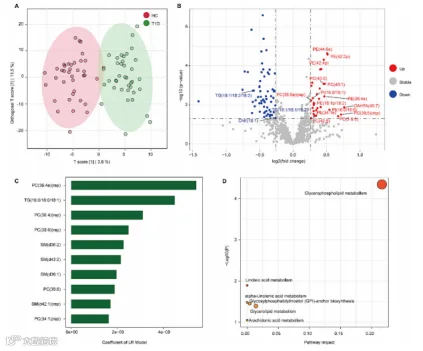
图3. T1D中脂质组特征的改变。
(A) OPLS-DA显示了脂质组成的区分趋势。
(B) 火山图显示了通过倍数变化(>1.2)和p值(<0.05)使用t检验的差异脂质筛选。
(C) 机器学习(ML)预测的对疾病有贡献的前10个脂质组特征。
(D) 差异脂质的代谢途径。
5、T1D相关的肠道微生物与血清代谢物的相关性
差异性血清代谢物和T1D相关的肠道微生物之间的关联分析显示在图4A中。结果表明,Ruminococcus torques与参与调节膜脂性质的3-羟基丁酸呈正相关(r = 0.474,p < 0.05)。O-乙酰基肉碱,也称为乙酰基肉碱,参与了重要的代谢过程,特别是能量转移,它与Lactobacillales(r = 0.343,p < 0.05)和Streptococcaceae(r = 0.346,p < 0.05)呈正相关。
还有其他的菌群和代谢物之间的关联,例如Slackia和酰胺C18之间的负相关;Flavonifractor和C22H43NO之间的负相关;Flavonifractor和C18H42N5P之间的负相关;(e)-巴尼酸、9-氧代-十八碳二烯酸、C16H26N6O2和Lachnospiraceae_UCG_001之间的正相关;以及(e)-巴尼酸、9-氧代-十八碳二烯酸、C16H26N6O2和Intestinibacter之间的负相关。
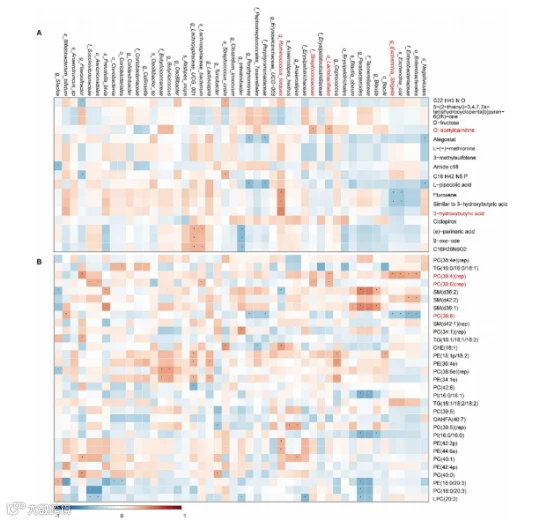
图4. T1D疾病特异性的相关性分析。
(A) T1D相关微生物和代谢物的相关性分析结果。*p<=0.05。
(B) T1D相关微生物和脂质的相关性分析结果。*p<=0.05。
6、T1D相关的肠道微生物与血清脂质的相关性
预测的脂质和T1D相关的肠道微生物之间的关联分析显示出多样的结果(图4B)。一些磷脂酰胆碱与微生物显示出显著的相关性。磷脂酰胆碱(36:4)(rep)与大肠埃希氏菌(r = 0.394,p < 0.05)呈正相关,并与乳杆菌目(r = 0.340,p < 0.05)呈正相关。磷脂酰胆碱(38:6)(rep)与乳杆菌目(r = 0.338,p < 0.05)和链球菌科(r = 0.340,p < 0.05)呈正相关。磷脂酰胆碱(38:6)与大肠埃希氏菌(r = 0.322,p < 0.05)呈正相关。
此外,还发现了菌群和脂质谱之间的其他关联。例如,大肠埃希氏菌、大肠杆菌、肠杆菌科和肠杆菌目与PC(36:4)(rep)呈正相关,但与PC(38:6)呈负相关。然而,拟杆菌属和坦纳菌科与PI(16:0/18:1)、PI(16:0/16:0)、PE(18:0/20:3)、PC(18:0/20:3)和LPC(20:3)呈负相关,而与SM(d36:2)和SM(d36:1)呈正相关。
讨 论
通过ML和LefSe筛选的T1D微生物特征
Ruminococcus torques、Anaerostipes、Veillonella、Erysipelotrichaceae UCG-003、Blautia和Coprococcus是通过ML和LefSe筛选出的重叠微生物(所有在T1D中增加)。一项研究报告说,Ruminococcus torques在HbA1c水平较高(53 mmol/mol)的T1D患者中增加。
此外,Akkermansia muciniphila已经被证明能够通过在微生物群中与Ruminococcus torques竞争来控制胰岛自身免疫和延缓NOD小鼠的糖尿病发展。Ruminococcus torques可能通过诱导炎症和破坏肠道屏障来促进自身免疫性疾病(例如T1D)的发展。Ruminococcus torques引起的炎症的主要机制之一可能是影响能够引起全身性炎症的细胞因子水平。
以前的一项研究表明,Ruminococcus torques与IL-10水平呈反相关,以及与可以诱导T2D大鼠全身性炎症的脂多糖水平呈正相关。在一种以腹泻为主的肠易激综合征模型中,Ruminococcus torques和Blautia与IL-6、TNF-a、IL-1b和IFN-c呈正相关,这些是Fuzi-Lizhong丸在抑制全身性炎症过程中的重要肠道微生物。
第二个主要机制是Ruminococcus torques降解了肠上皮细胞表面粘液层中的粘蛋白,使细菌增殖,这可能是肠道屏障被破坏的一个重要原因,伴随着肠道炎症。
通过ML、WGCNA和倍数变化筛选的T1D代谢物特征
经过ML、WGCNA和倍数变化筛选,有七种重叠代谢物,分别是(e)-巴利纳酸、3-羟基丁酸、9-氧代-ODE、C16H26N6O2、西克洛匹罗、氟氧烯和一种类似于3-羟基丁酸的代谢物。在这些重叠代谢物中,3-羟基丁酸和9-氧代-ODE已被报道与糖尿病或相关的代谢表型有关。9-氧代-ODE是一种氧化亚油酸代谢物,在我们的研究中,T1D患者中增加了。在以前的研究中,9-氧代-ODE在有代谢综合征的人类中也有更高的水平,而在没有代谢综合征的人类中则没有。9-氧代-ODE的升高与代谢综合征患者肠道细菌负荷的降低有关。
与HCs相比,我们的研究中T1D患者的代谢产物3-羟基丁酸增加了。3-羟基丁酸是最突出的酮体,它可以在某些葡萄糖缺乏或吸收不良的情况下提供能量。它也是有毒的,导致T1D患者酮症酸中毒。
此外,发现持续的轻度酮血症会增加低密度脂蛋白胆固醇水平,这可能加剧心血管疾病的危险。然而,有证据表明3-羟基丁酸对代谢和心血管疾病有益。
对于低血糖的人,3-羟基丁酸是一种抵御胰岛素诱导的低血糖的防御。此外,3-羟基丁酸抑制了脂肪分解、炎症、氧化损伤、癌症发展、血管生成和动脉粥样硬化,可能通过体力活动和热量限制帮助延长寿命。
经过机器学习(ML)和倍数变化筛选的T1D的脂质特征
在我们的研究中,T1D患者中SM(d36:1)、TG(18:1/18:1/18:2)、ChE(18:1)和TG(18:1/18:2/18:2)降低了,而TG(16:0/16:0/18:1)没有降低。ML和10种重叠脂质的前10种血清脂质在T1D患者中增加了。PC(36:4)(rep)在T1D患者中增加了。
在以前的研究中,这种脂质在与抗体阴性受试者相比,抗体阳性的糖尿病受试者中增加了,与新发T1D患者中不同的C-肽水平有明显的关系。PC(36:4)也被发现与红肉摄入和糖尿病风险有关。PC(38:6)、TG(16:0/16:0/18:1)和TG(18:1/18:2/18:2)在以前的研究中也显示出与T1D有正或负的关联。
鞘磷脂在T1D的进展中可以发生关键性的改变。T1D患者的血清脂质组通过改变的鞘磷脂水平来识别,例如与我们的研究中的HCs相比,SM(d36:1)降低,而SM(d36:2)、SM(d42:2)和SM(d42:1)(rep)增加。在T1D患者中,当他们遵循低碳水化合物饮食时,SM(d36:1)显著增加。以前的数据表明,鞘磷脂在T1D发展过程中的β细胞死亡中起着至关重要的作用。此外,以前的研究已经证实了鞘磷脂衍生的神经酰胺在肝脏的胰岛素抵抗和白蛋白尿的发展以及药理学抑制葡萄糖稳态中的作用。
微生物组和代谢组以及脂质组在T1D中的关系
在我们的研究中,Ruminococcus torques在T1D患者中增加,并且与HbA1c水平呈正相关,与以前的研究一致。此外,3-羟基丁酸,这种在本研究中T1D患者中增加的代谢物,参与了酮体的合成和降解以及丁酸代谢。
另外,Ruminococcus torques与T1D患者中的3-羟基丁酸水平呈正相关(p < 0.01,r = 0.474)。Ruminococcus torques本身也是一种产丁酸的细菌物种。Ruminococcus torques可以发酵膳食纤维来产生丁酸。丁酸是结肠细胞的主要能量来源,丁酸被代谢成酮体。
此外,在我们的研究中,大肠杆菌志贺氏菌与T1D患者中的PC(36:4)水平呈正相关。一些研究报道,大肠杆菌志贺氏菌可以通过干扰宿主的脂质代谢影响代谢性疾病的发展。在我们的研究中,一些磷脂酰胆碱(如PC(36:4)(rep)和PC(38:6)(rep))和O-乙酰肉碱在T1D患者中增加,它们是一种名为三甲胺-N-氧化物(TMAO)的肠道来源的代谢物的前体,它也与心血管疾病和糖尿病有关。胆碱转化为TMAO需要胆碱三甲胺裂解酶,该酶由三种肠道细菌门(厚壁菌门、变形菌门和放线菌门)编码。大肠杆菌志贺氏菌属于变形菌门。
此外,我们还发现乳杆菌目---PC(38:6)(rep)、链球菌科---PC(38:6)(rep)、乳杆菌目---PC(36:4)(rep)、乳杆菌目---O-乙酰肉碱和链球菌科---O-乙酰肉碱之间存在正相关。乳杆菌目和链球菌科也属于编码三甲胺裂解酶的细菌门,在我们的研究中,它们与HbA1c水平呈正相关。
T1D中组学特征的实际应用
T1D患者中组学特征及其关系的发现为未来的精准诊断和治疗提供了潜在的靶点。目前的研究已经探索了使用疾病特异性益生菌和微生物群移植作为阻止T1D进展的策略。此外,抗生素耐药材料的开发,如纳米复合材料,有望作为一种有效的方法来改善肠道微生物群。以前的研究已经报道,DyBa2Fe3O7.988/DyFeO3纳米复合材料可以作为抗菌剂,因为它们对革兰氏阴性病原体,如肺炎克雷伯菌和大肠杆菌,具有强大的抗菌活性。
另一项研究强调,Co/Co3O4纳米复合材料对革兰氏阴性微生物,包括铜绿假单胞菌,表现出强大的抗菌活性。
这些发现共同强调了针对微生物群和生物标志物治疗疾病的新兴技术的有利前景。
结论
在本研究中,我们结合了机器学习和传统的生物统计学,来识别T1D的微生物、代谢组和脂质组特征,并进一步探讨了T1D相关的微生物群与相关代谢物和脂质代谢在T1D进展中的关系。
我们研究的优点是参与者的特征明确,多组学的目标和定量性,以及机器学习和其他生物统计学的结合使用。
这项研究也有一些局限性,包括相对较小的样本量和缺乏前瞻性研究来验证筛选出的特征对T1D进展的影响。
本文译自:
Tan Huiling, Shi Yu, Yue Tong, et al. Machine learning approach reveals microbiome, metabolome, and lipidome profiles in type 1 diabetes. J Adv Res. 2023 Nov 30: S2090-1232 (23)00363-6.
往期推荐
