
奇辉观点
炎症性肠病(IBD)的全球发病率和患病率正在逐渐增加。高脂饮食(HFD)已知会破坏肠道稳态并加剧IBD,但背后的机制尚未完全明确。在本研究中,观察到饮食脂肪摄入量与IBD患者和鼠类结肠炎模型疾病严重程度之间的正相关性。高脂饮食导致吲哚-3-乙酸(IAA)显著减少,并导致肠道屏障损伤。此外,IAA补充增强了肠道黏液硫酸化,并有效缓解了结肠炎。在机制上,IAA通过芳烃受体(AHR)上调黏液硫酸化的关键分子,包括3'-磷酸腺苷5'-磷酸硫酸合酶2(Papss2)和溶质载体家族35成员B3(Slc35b3),这是3'-磷酸腺苷-5'-磷酸硫酸(PAPS)的合成酶和转移酶。更重要的是,AHR可以直接结合到Papss2的转录起始位点。口服能够产生IAA的罗伊氏乳杆菌有助于保护肠道免受结肠炎的侵害并促进黏液硫酸化,而缺乏iaaM基因(LactobacillusΔiaaM)和产生IAA能力的改良罗伊氏乳杆菌株未能显示出这种效果。总体而言,IAA通过AHR-Papss2-Slc35b3途径增强肠道黏液硫酸化,有助于保护肠道稳态。研究结果强调了饮食,特别是高脂饮食,对肠道微生物群落和肠道健康的影响,为通过饮食干预来预防或治疗IBD提供了依据;通过补充IAA或利用能够产生IAA的益生菌,可能为IBD的治疗提供新的方法。
论文ID
本文译自:Li M, Ding Y, Wei J, et al. Gut microbiota metabolite indole-3-acetic acid maintains intestinal epithelial homeostasis through mucin sulfation. Gut Microbes. 2024 Jan-Dec; 16(1): 2377576.
发表杂志:GUT MICROBES
影响因子:12.3
通讯作者:Hailong Cao
作者单位:Tianjin Medical University General Hospital
引 言
炎症性肠病(IBD)近几十年来在全球范围内的患病率有所增加,但其病因仍然不明。IBD的发病与环境因素(如西方饮食方式)、遗传易感性、肠道微生物群落组成的干扰以及肠道屏障完整性受损有关。宏基因组学和代谢组学分析揭示了IBD患者肠道微生物群落及其代谢产物的显著失衡,特征是物种多样性减少、厚壁菌门的丰度降低以及肠杆菌科的增加。肠道微生物的代谢产物,如短链脂肪酸、次级胆汁酸和色氨酸衍生物,都有所减少。流行病学研究表明,西方饮食方式的全球兴起与IBD发病率的区域性增加之间存在同步性,并且西方饮食方式与IBD进展的风险增加显著相关。基于大豆油的高脂饮食(HFD)通过减少抗炎微生物代谢产物,增加了IL-10基因敲除小鼠对结肠炎的易感性,而45%的脂肪饮食与抗生素联合使用,协同损害肠道上皮的线粒体功能,引发肠道微生物群落的失衡并加剧粘膜炎症。然而,高脂饮食暴露与肠道稳态紊乱之间的联系仍然不完全清楚。
色氨酸是人体必需的芳香族氨基酸,可以被肠道内的肠道微生物进一步处理。微生物衍生的色氨酸代谢产物,如吲哚、吲哚-3-醛(I3A)、吲哚乙醛和吲哚乳酸(ILA),因其作为芳烃受体(AHR)的配体而对宿主生理产生显著影响。AHR是一种广泛保守的配体激活转录因子,在没有配体激活的情况下位于细胞质中。配体结合后,它转移到细胞核并与AHR核转运蛋白(Arnt)形成复合物。这个复合物随后结合到DNA上的异生物质响应元件(XREs),从而调节目标基因的转录。由大肠杆菌等细菌产生的ILA通过AHR下调上皮CCL2/7的产生,从而减轻巨噬细胞的激活。此外,I3A、吲哚-3-丙酮酸和吲哚-3-乙醇,这三种来自肠道微生物的色氨酸代谢产物,在右旋糖酐硫酸钠(DSS)诱导的结肠炎期间显示出保护肠道上皮屏障完整性的潜力。然而,其他色氨酸衍生物在维持肠道稳态方面的影响和潜在机制尚未被研究。临床证据表明,IBD患者在炎症组织中表现出AHR的表达减少和活性降低,这种现象可能与色氨酸代谢产物的减少有关。
粘液层位于大肠微生物群和上皮及免疫细胞之间,对于保持肠腔内容物(包括细菌)与上皮细胞的隔离起着至关重要的作用。粘液层主要由高度糖基化的黏液素2(MUC2)组成,进一步分为酸性和中性黏液,以及硫酸黏液和唾液酸黏液的亚型。硫酸黏液是O-糖基化修饰的一种普遍形式,在小肠和结肠中都很常见,而在远端结肠中观察到相对较高水平。远端结肠中硫酸化黏液水平的增加有助于阻碍细菌对黏液糖链的酶解,从而有效防止细菌、有害因素与肠道上皮的密切接触。
在这项研究中,我们观察到高脂饮食(HFD)破坏了小鼠肠道中的色氨酸代谢,导致吲哚-3-乙酸(IAA)水平显著下降。随后补充IAA和能够产生IAA的罗伊氏乳杆菌(Lactobacillus reuteri)能够缓解小鼠的结肠炎并增厚结肠粘液层。在机制上,IAA通过芳烃受体(AHR)上调了黏液硫酸化过程中的关键分子3'-磷酸腺苷5'-磷酸硫酸合酶2(Papss2)和溶质载体家族35成员B3(Slc35b3)的表达,从而增强肠道屏障功能并改善结肠炎。
结 果
饮食脂肪加剧了DSS诱导的结肠炎
为了揭示高脂饮食(HFD)对患有UC或CD患者的肠道稳态的影响,我们对87名UC患者和50名CD患者进行了回顾性饮食调查。我们的发现显示,UC和CD患者的饮食脂肪摄入量与疾病活动指标之间存在正相关,如UC患者的Mayo评分(R = 0.3183, p < 0.0001)、粪便钙卫蛋白水平(R = 0.2417, p < 0.0005)和ESR(R = 0.2417, p < 0.0001),CD患者的克罗恩病活动指数(R = 0.2734, p < 0.0001)、粪便钙卫蛋白水平(R = 0.1685, p = 0.0042)和ESR(R = 0.1114, p = 0.0112)(图1a)。此外,结肠镜和组织病理学图像表明,采用HFD的UC患者可能会发展出更严重的炎症和粘膜损伤(图1b)。
为了检验饮食脂肪对结肠炎的影响,我们首先让小鼠分别进食HFD或普通饮食2周,然后在HFD喂养的小鼠和普通饮食喂养的小鼠体重差异显著出现之前给予2.5% DSS(图1c)。在收获时,喂养HFD的小鼠比普通饮食喂养的小鼠有更多的DSS诱导的体重损失(图1d)。此外,DAI评分显示,喂养HFD的小鼠对DSS处理更敏感,患有更严重的侵袭性结肠炎(图1e)。喂养HFD的小鼠的结肠比对照组短,并且显示出更小的盲肠和明显的血性侵蚀(图1f)。组织病理学揭示了结肠炎的典型特征,包括减少的上皮隐窝密度或增加的隐窝间隙,以及广泛的炎症细胞浸润和溃疡的存在。HFD加剧了这些现象,导致炎症细胞浸润增加和隐窝消失,与普通饮食相比,组织病理学评分增加(图1g)。同时,HFD增加了肠道上皮通透性,导致细菌易位增加(图1h)。总的来说,这些发现表明短期HFD加剧了DSS诱导的结肠炎并导致肠道屏障损伤。
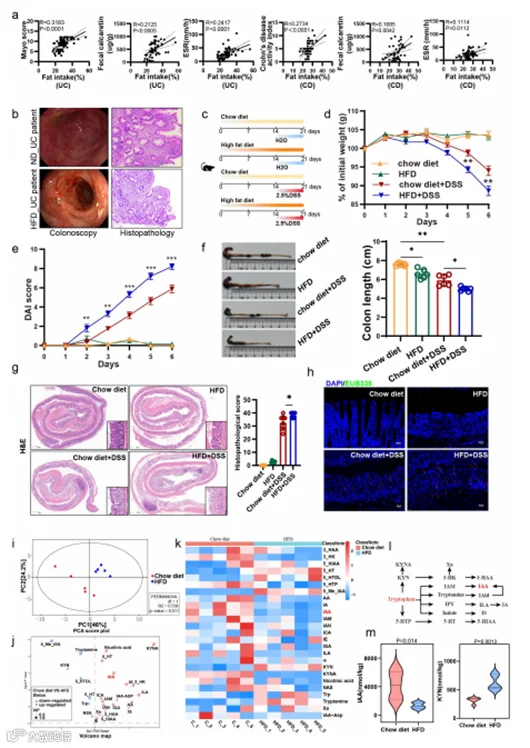
图1. 高脂饮食加剧了小鼠的结肠炎并诱导了肠道色氨酸代谢的紊乱。
肠道代谢组揭示了喂养高脂饮食的小鼠体内IAA的显著减少
一些研究表明,高脂饮食(HFD)可以改变肠道微生物群的组成,随后改变微生物的代谢特征。为了更深入地探讨HFD与色氨酸代谢之间的关系,对喂养普通饮食和HFD的小鼠在喂养2周后的粪便样本进行了针对性的色氨酸代谢组分析。结果,通过主成分分析(PCA)观察到喂养普通饮食的小鼠和喂养HFD的小鼠之间存在部分但显著的分离(图1i)。在喂养HFD的小鼠中,分别有7个和18个模块显著富集或减少(图1j)。其中,色氨酸代谢途径中的犬尿氨酸(kyn)和血清素途径的代谢产物在HFD小鼠的粪便中增加,例如kyn,除了犬尿氨酸(KYNA),已被证明通过增强肠道上皮细胞的增殖来减轻结肠炎的严重程度。不幸的是,对于肠道稳态至关重要的吲哚途径的吲哚类化合物在HFD小鼠的粪便中普遍减少,包括IAA和吲哚丙烯酸(IA)(图1k-m)。
IAA减轻了结肠炎的严重程度并增强了肠道屏障功能
IAA与结肠炎之间的关联仍然相对未被探索。为了阐明IAA在结肠炎中的潜在作用,我们在实验结束前7天开始,通过口服方式将IAA给予DSS诱导的急性结肠炎小鼠(图2a)。我们发现,与接受水的对照组相比,DSS给药成功地导致小鼠明显的体重损失。尽管如此,给小鼠施用IAA显著减轻了DSS诱导的体重损失(图2b)。为了评估结肠炎的严重程度,我们在建模期间每天监测DAI评分。结果,DSS显著增加了DAI评分,而基于DSS的IAA补充显著降低了DAI评分,表明IAA可以缓解结肠炎的严重程度(图2c)。同时,在安乐死小鼠之前,内窥镜检查显示DSS给药导致肠道腔内广泛的粘膜侵蚀和出血,在某些情况下观察到粘膜脱落,而IAA减少了溃疡面积和出血程度(图2e)。一致地,IAA也显著缓解了接受DSS挑战的小鼠观察到的结肠长度减少(图2d)。更重要的是,与单独给予DSS相比,IAA补充增加了隐窝数量和隐窝高度,减少了炎症细胞的浸润,并减轻了上皮细胞损伤,并显著降低了病理评分(图2f)。我们进一步评估了IAA对炎症细胞因子的影响,发现IAA喂养显著降低了DSS处理的小鼠结肠组织中IL-1β、IL-6和TNFα的水平(图2g)。免疫荧光分析显示,与单独DSS处理相比,同时接受IAA治疗的结肠炎模型小鼠ZO-1表达增加(图2h-i)。此外,DSS处理的小鼠显示细菌渗透到上皮细胞的增加,而IAA减少了细菌向肠道上皮的易位(图2j)。
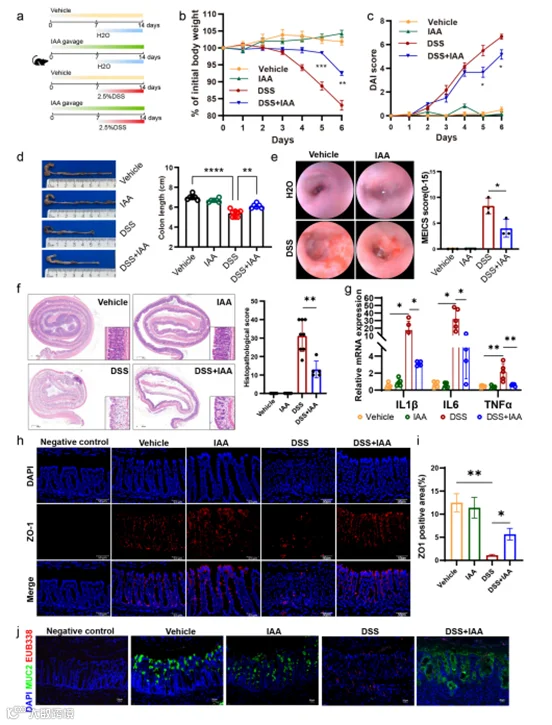
图2. IAA减轻了DSS诱导的结肠炎的严重程度并增强了肠道屏障功能。
为了进一步阐明IAA的效果,我们使用TNBS诱导结肠炎并观察到类似的发现。IAA给药降低了小鼠的死亡率、体重损失、结肠缩短,并缓解了TNBS诱导的粘膜上皮损伤(图S2a-d)。此外,接受IAA治疗的结肠炎小鼠表现出较低水平的促炎细胞因子,包括IL-1β和TNFα(图S2e),与结肠炎小鼠相比。为了更好地评估IAA在结肠炎中的治疗潜力,并减轻IAA与DSS药理效应的潜在干扰,我们最初让小鼠饮用含有DSS的水7天。随后,在出现明显的结肠炎症状后,小鼠在第8天接受IAA治疗5天(图S3a)。结果表明,IAA组的小鼠肠道损伤恢复更快,肠道通透性更低,炎症减轻,与对照组相比(图S3b-k)。总的来说,这些数据证明了IAA治疗可以缓解结肠炎模型小鼠的结肠炎症并恢复上皮损伤。
IAA通过促进黏液硫酸化增强了粘膜屏障功能
在体内实验中,我们进一步观察到,与DSS处理组相比,补充IAA的小鼠的杯状细胞数量和粘液层厚度显著增加(图4a-b,图S1a-b)。为了进一步挖掘IAA处理小鼠结肠粘液层改善背后的机制,我们对DSS处理的小鼠是否补充IAA的结肠组织进行了RNA测序分析。结果表明,两组之间的转录组存在显著差异(图3a-b)。在DSS处理的小鼠中添加IAA抑制了如IL-6、IL-1β和Cxcl2等炎症细胞因子的基因表达(图S4b)。此外,使用京都基因与基因组百科全书(KEGG)途径分析差异表达基因显示,多个信号途径显著富集。其中,硫代谢信号显著富集(图S4a)。基因集富集分析(GSEA)显示IAA处理改变了细胞质硫酸化途径(图3c,d)。硫酸化过程由硫酸转移酶促进,是一个关键的结合反应,硫酸化碳水化合物构成了黏液成分的重要部分。硫酸化通常以末端帽的形式出现在黏液中,通常防止寡糖酶解。3'-磷酸腺苷5'-磷酸硫酸(PAPS)是硫酸供体,Papss2是催化PAPS形成的的关键酶。然后,PAPS转运蛋白Slc35b3负责将PAPS运输到高尔基体参与黏液硫酸化反应。据报道,Papss2的肠道缺乏会损害黏液硫酸化和肠道屏障在结肠炎挑战时的功能。在我们的研究中,RNA测序数据显示,IAA处理的结肠炎小鼠中Papss2和Slc35b3的表达显著增加(图3d)。此外,我们对未补充IAA但未进行DSS处理的小鼠进行了RNA测序分析,观察到这两组之间的转录组存在显著差异(图S4c)。然而,我们没有观察到细胞质硫酸化途径的富集。尽管如此,我们确实发现IAA组中Papss2的mRNA表达增加(图S4d)。结果通过RT-PCR得到确认(图4e)。
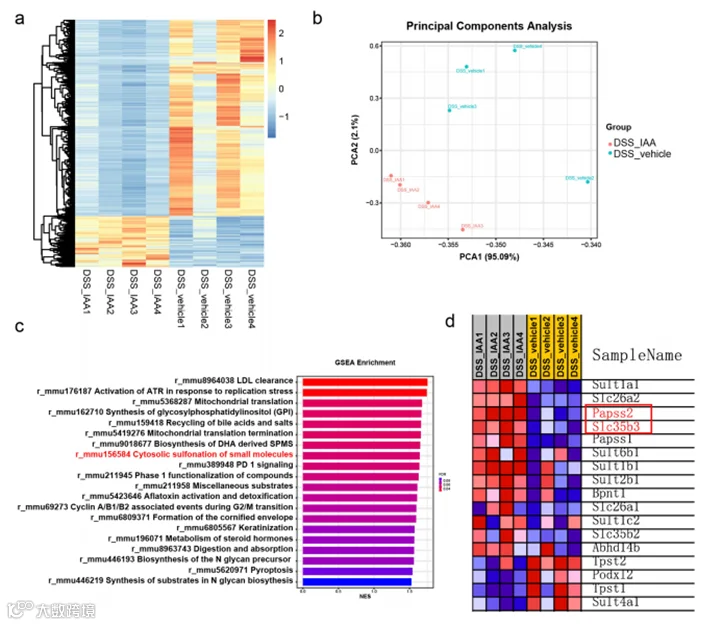
图3. 转录组分析揭示IAA促进了硫酸化途径的富集。
HID-AB染色和凝集素MALII染色允许特定可视化黏液中硫酸黏液亚型。在我们的研究中,我们观察到与未处理的小鼠相比,DSS处理的小鼠结肠组织中硫酸化黏液显著减少,而IAA增加了结肠上皮中硫酸化黏液的含量,特别是在远端结肠(图4c,d;图S1c-e)。此外,我们观察到与对照组相比,DSS组中Papss2和Slc35b3的表达减少,而基于DSS给药的IAA补充恢复了它们的表达,正如我们之前提到的(图4e)。一致地,IAA处理恢复了DSS处理的结肠组织中Papss2的蛋白表达(图4f-g)。我们还发现IAA促进了GlcNAc6st2的表达(图4e),据报道,GlcNAc6st2是小鼠结肠黏液硫酸化修饰中的主要硫酸转移酶。
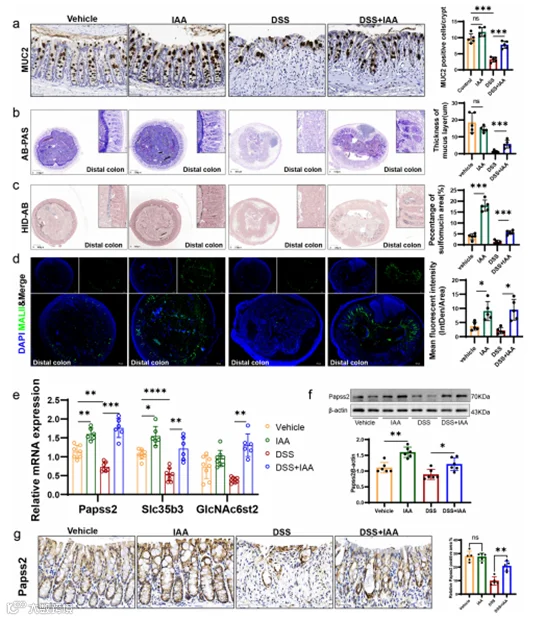
图4. IAA补充通过促进黏液硫酸化增强了粘膜屏障功能。
在确定并验证IL-10-/-小鼠的基因型(图5a)的基础上。我们补充了11周龄的IL-10−/−小鼠是否补充IAA,并观察到当小鼠达到16周时,对照组发展出显著的自发性结肠炎,而IAA组没有表现出明显的炎症。具体来说,与对照组相比,IAA组的DAI评分较低,体重损失较少,结肠缩短较少,组织病理学评分较低,炎症细胞因子水平较低(图5b-f)。此外,使用HID-AB染色和MALII凝集素染色评估两组中硫酸化黏液的水平,我们发现IAA组在近端、中部或远端结肠中硫酸化黏液的水平较高(图5i,j;图S5a-d)。此外,IAA补充促进了Papss2、Slc35b3和GlcNAc6st2的mRNA表达(图5g),以及Papss2的蛋白表达(图5h)。
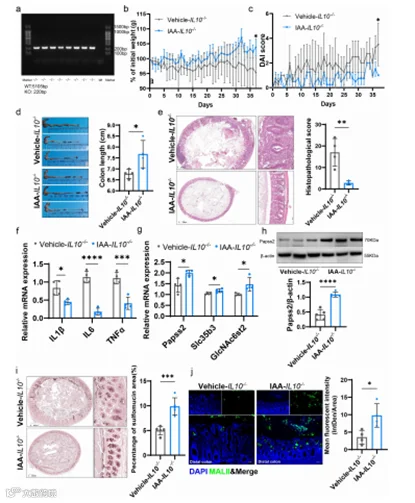
图5. IAA补充改善了IL10-/-小鼠的自发性结肠炎并促进了黏液硫酸化。
IAA对黏液硫酸化的影响部分依赖于AHR
结肠中的色氨酸可以被肠道微生物代谢成一系列吲哚代谢产物,包括IAA。已有报道称IAA作为AHR或PXR的配体发挥作用。为了验证哪个受体在IAA介导的Papss2表达增加中起作用,我们在体外用PXR和AHR抑制剂预处理LS174T和HT29细胞,然后再进行LPS和IAA处理。然而,我们发现只有AHR抑制剂显著抑制了Papss2的表达,在LPS+IAA和LPS+IAA+PXRi组之间没有观察到统计学上的显著差异(图6a,b)。因此,我们得出结论,AHR在诱导Papss2表达中起主导作用。分子对接分析显示IAA与AHR的配体结合域结合,结合能为-5.246 kcal/mol,这表明IAA具有显著的结合AHR的能力(图6c)。此外,我们观察到细胞同时暴露于LPS和IAA后,AHR的核转位增加(图6d)。这些数据表明IAA能够结合AHR,促进AHR复合物进入细胞核,然后调节目标基因的表达。
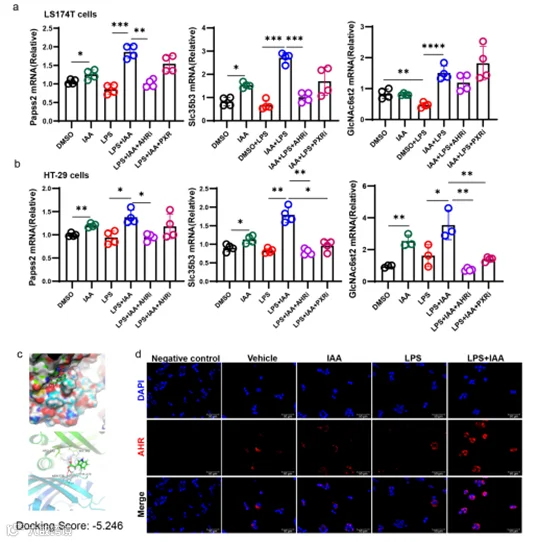
图6. IAA激活AHR以调节黏液硫酸化。
为了进一步研究IAA对肠道黏液硫酸化的影响是否通过AHR的介导来协调,我们给Ahr−/−小鼠施用IAA,然后进行DSS挑战(图7a)。结果表明,在Ahr−/−小鼠中,IAA缓解了DSS诱导的体重损失,但并未改善存活率、DAI评分、结肠缩短或组织病理学评分(图7b-e)。我们得出结论,在Ahr−/−小鼠中补充IAA并未改善DSS引起的肠道上皮损伤(图7f)。HID-AB染色揭示了AHR基因敲除抵消了IAA诱导的硫酸黏液水平的升高(图7g)。至关重要的是,我们的研究包括了肠道组织的定量RT-PCR,揭示了IAA给药逆转了DSS在体内引起的Papss2和Slc35b3水平的降低,这种效应在Ahr−/−小鼠中减弱(图8a)。一致地,Papss2的蛋白表达水平也显示出类似的结果(图8b,c)。
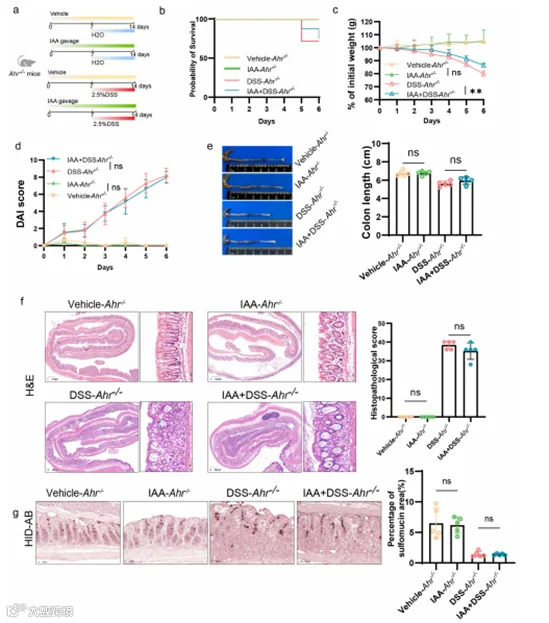
图7. IAA对黏液硫酸化的影响部分依赖于AHR。
IAA通过AHR-XRE与Papss2启动子的相互作用增加Papss2表达
作为转录因子,AHR在结合配体后转移到细胞核,然后结合到DNA上调节目标基因的转录。在体内和体外实验中,我们发现AHR抑制剂或AHR基因敲除部分消除了IAA对Papss2基因表达的促进作用。因此,我们假设Papss2可能是AHR的靶基因。进一步采用Cut&tag实验,我们发现AHR在Papss2和Slc35b3的基因启动子区域富集。这表明AHR可以直接调节Papss2和Slc35b3基因的转录(图8d)。我们进一步分析了AHR在Papss2和Slc35b3启动子上的结合位点,并发现Papss2和Slc35b3启动子区域都存在XRE(TGCGTG/CGCGTG),与JASPAR数据库中AHR的结合位点一致(图8e)。这些结果表明,AHR通过结合Papss2和Slc35b3启动子区域的XREs直接调节这两个基因的转录,并进一步调节硫酸化过程。
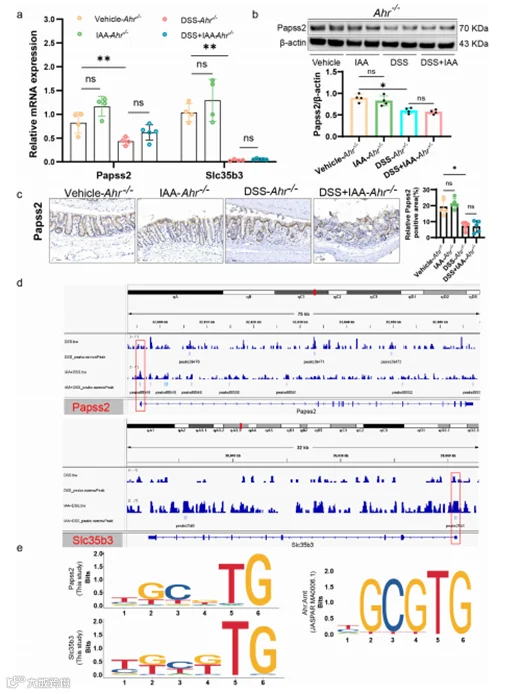
图8. AHR-XRE相互作用介导了IAA对Papss2的影响。
罗伊氏乳杆菌衍生的IAA是缓解结肠炎和促进黏液硫酸化所必需且足够的
罗伊氏乳杆菌已被证明能够产生包括IAA和I3A在内的多种吲哚衍生物。为了进一步阐明IAA在改善结肠炎中的主要作用,我们构建了一个缺乏iaaM基因的罗伊氏乳杆菌突变株(LactobacillusΔiaaM),导致无法产生IAA。我们在体外评估了罗伊氏乳杆菌和LactobacillusΔiaaM的IAA生产能力(图S6a)。
随后,我们对C57BL/6J小鼠进行了PBS、罗伊氏乳杆菌或LactobacillusΔiaaM的口服灌胃。这些小鼠在2周前用抗生素混合物(ABX)处理以耗尽肠道微生物群(图9a)。在确认罗伊氏乳杆菌和LactobacillusΔiaaM成功定植后(图S6b),我们在最后12天开始同时进行DSS处理。结果显示,罗伊氏乳杆菌减轻了DSS引起的体重增加、DAI评分高和结肠长度缩短,并且与单独DSS处理相比,罗伊氏乳杆菌灌胃组的小鼠炎症有显著改善(图9b-e)。然而,LactobacillusΔiaaM灌胃组与DSS组相比并没有显著差异。HID-AB染色揭示了与PBS组相比,口服罗伊氏乳杆菌后结肠黏液硫酸化含量在杯状细胞中显著增加。然而,与罗伊氏乳杆菌相比,LactobacillusΔiaaM在促进黏液硫酸化方面的能力显著降低(图9f)。上述结果表明,IAA在保护罗伊氏乳杆菌抵御结肠炎和促进黏液硫酸化方面是必不可少的。
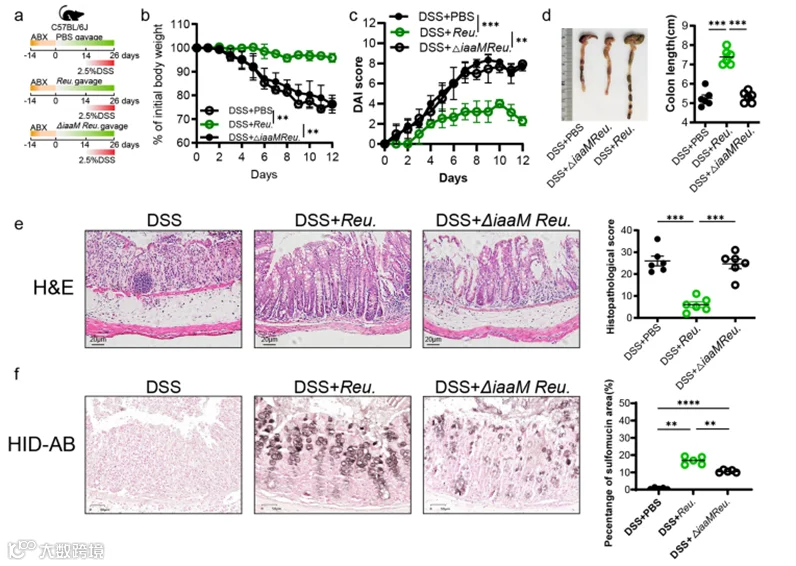
图9. 罗伊氏乳杆菌衍生的IAA是缓解结肠炎和促进黏液硫酸化所必需且足够的。
暴露于高脂饮食的UC患者Papss2表达和黏液硫酸化水平下调
由于Papss2的表达和黏液硫酸化水平在小鼠结肠炎中表现出减少的表达,探索IBD患者中的表达概况具有重要的意义。对基因表达综合数据库(GEO)中结肠组织转录组数据(GSE59071)的分析表明,与健康个体相比,UC和CD患者的Papss2和Slc35b3表达显著降低(图10a)。来自左侧UC患者降结肠炎症组织和升结肠非炎症组织的单细胞RNA测序揭示了与非炎症组织相比,炎症组织中杯状细胞内Papss2和Slc35b3的表达降低(图10b,c),这与GEO数据库一致,表明杯状细胞黏液硫酸化的减少可能与炎症的发生密切相关。在前面的饮食调查中,确定了高脂饮食与IBD疾病活动之间的关联。最后,我们对UC患者和暴露于高脂饮食的UC患者的结肠组织切片进行了HID-AB染色和Papss2的免疫组化染色。结果显示,暴露于高脂饮食的UC患者黏液硫酸化水平和Papss2的表达明显低于UC患者(图10d,e)。总之,上述结果表明Papss2和硫酸化与IBD以及高脂饮食之间存在密切关联。
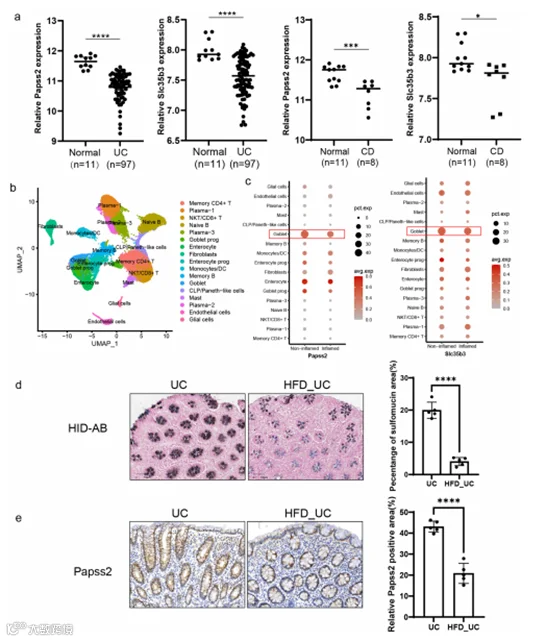
图10. 暴露于高脂饮食的UC患者Papss2表达和黏液硫酸化水平下调
讨 论
越来越多的证据表明,西方的高脂饮食(HFD)影响IBD的风险。在我们的研究中,我们也证明了高脂饮食可能加剧结肠炎的严重程度,并破坏DSS诱导的结肠炎模型中的肠道屏障功能。以前的研究已经揭示了高脂饮食可能塑造活性组织(如免疫细胞和脂肪细胞)的代谢。为了预防饮食引起的代谢变化对肠道炎症的潜在影响,我们专门给小鼠进行了为期2周的高脂饮食。在DSS诱导的结肠炎建模之前,与普通饮食的比较分析表明,高脂饮食并未引起小鼠体重的显著波动,这排除了高脂饮食通过改变细胞代谢影响结肠炎的可能性。一些研究报告称,高脂饮食耗尽了某些肠道代谢物的水平,然后增加了宿主对病原体的易感性。在我们之前的实验中,我们发现母体高脂饮食可能通过改变肠道微生物色氨酸代谢并降低微生物相关的吲哚代谢物水平,破坏了后代的肠道黏液屏障。我们进一步研究了高脂饮食对色氨酸代谢的具体影响,也发现与普通饮食喂养的小鼠相比,高脂饮食喂养的小鼠显示出独特的色氨酸代谢组特征。具体来说,高脂饮食增加了粪便样本中犬尿氨酸(Kyn)的水平,并降低了吲哚乙酸(IAA)的水平。这一结果得到了在高脂饮食诱导的代谢综合征和非酒精性脂肪肝病小鼠模型中报告的发现的支持,其中小鼠分别被喂养高脂饮食12周和8周。值得注意的是,IBD患者和肥胖受试者中也观察到Kyn水平更高和IAA水平更低。
最近的一项研究发现,高脂饮食可能破坏血清色氨酸代谢并上调犬尿氨酸代谢途径, 这与我们的发现一致。一些微生物衍生的色氨酸代谢物已被报道在减少结肠炎症和保护肠道上皮完整性方面发挥重要作用。因此,我们提出假设,高脂饮食失调肠道色氨酸代谢,导致IAA水平下降,这反过来剥夺了IAA对高脂饮食诱导的炎症和肠道屏障功能破坏的保护作用。我们随后的体内实验验证了IAA可以显著缓解结肠炎并保护肠道屏障完整性。这一点进一步得到了RNA测序分析结果的支持。与对照结肠炎动物相比,接受IAA治疗的小鼠中,如IL-1β、IL-4、IL-6和Cxcl2等炎症标志物的表达显著降低。在机制上,我们观察到DSS加IAA组中细胞质硫酸化途径富集,其中Papss2和Slc35b3的表达显著升高。这两个基因在黏液硫酸化修饰过程中都是必不可少的。在结肠中,O-糖链通过硫酸化广泛修饰,以阻止细菌对糖链的酶解。编码硫酸酯酶的细菌可以识别硫酸化糖并去硫酸化它们,随后分解黏液糖链以获取能量来源。临床调查表明,IBD患者的粪便黏液中硫酸酯酶活性升高。相应地,黏液硫酸化水平的降低可能会促进黏液降解细菌(如脆弱拟杆菌)在黏液层中的增殖,从而促进它们的定植和生长。这种增加的细菌易位可能会反过来在易感宿主中引发炎症反应。肠道黏液屏障的功能在很大程度上取决于黏液的硫酸化。缺乏钠硫酸盐共转运体1(NaS1)的小鼠表现出降低的黏液硫酸化,并增加了对实验性结肠炎的易感性。47 黏液的硫酸化涉及硫酸转移酶将SO3−从PAPS转移到黏液的O-糖链。PAPS是体内普遍的硫酸化供体,由Papss1和Papss2合成。传统上,人们认为Papss1主要定位于各种细胞类型的细胞核,而Papss2主要位于细胞质。与这些观点相反,我们的免疫组化分析揭示了在我们的实验设置中细胞核内存在Papss2,需要进一步研究其特定功能。Papss2缺乏的小鼠显示出降低的肠道硫酸黏液含量,并对DSS诱导的结肠炎高度敏感。此外,Papss2表达的降低与结直肠癌患者的生存率较差相关,表明其在IBD之外的潜在重要性。此外,Papss2缺陷与骨骼发育异常有关,强调了识别Papss2的调节靶点的重要性,不仅在IBD的背景下,还在与结直肠癌和骨骼发育相关的疾病中。此外,在黏液中,SO3−可能附着在N-乙酰-D-葡萄糖胺(6S-GlcNAc)和末端D-半乳糖(Gal)糖的6-羟基上,以及羟基位置3、4或6(分别为3S-Gal、4S-Gal和6S-Gal)。与GlcNAc6st1相比,GlcNAc6st2被报道是小鼠结肠硫酸化黏液生物合成中的主要硫酸转移酶。GlcNAc6st2的缺乏大大增加了DSS诱导的结肠炎中结肠的白细胞浸润,而丁酸可以诱导结肠上皮细胞中GlcNAc6st2的表达。在我们的工作中,我们还检查了GlcNAc6st2的表达,并发现IAA同样可以增强GlcNAc6st2的表达,可能是通过直接或间接机制。这些发现强调了调控黏液硫酸化的复杂调控途径,强调了环境因素(如丁酸和IAA)在调节结肠上皮细胞中GlcNAc6st2表达中的潜在相互作用。
传统上,IAA被认为是由罗伊氏乳杆菌合成的产物。酶iaaM在色氨酸转化为IAA的过程中至关重要,被设计为该途径中的关键限速步骤。在我们的研究中,我们产生了一个iaaM基因缺陷的罗伊氏乳杆菌菌株,并观察到野生型罗伊氏乳杆菌促进了黏液硫酸化,而iaaM缺陷菌株(LactobacillusΔiaaM)在增强黏液硫酸化方面的能力显著降低。我们的发现强调了IAA在调节黏液硫酸化中的关键作用。值得注意的是,长期以来的观察表明UC患者的黏液硫酸化减少, 然而,机制上的见解仍然难以捉摸。因此,识别改善黏液硫酸化的相应靶点是非常期待的。我们的研究不仅阐明了色氨酸代谢物和黏液硫酸化之间的联系,还研究了潜在的机制。与色氨酸相关的吲哚代谢物已报道其有潜力改善肠道屏障功能。因此,我们随后的努力涉及修改IAA,以实现在结肠中的目标释放,目标是提高其在缓解结肠炎中的治疗效果。这将为IAA在管理IBD中的临床实施奠定更加坚实的基础。
总的来说,这些发现强调了结肠黏液硫酸化在维持结肠黏膜稳态中的关键作用,并确定了IBD的潜在治疗靶点。然而,很少有研究阐明这些靶点的确切调控机制。在本研究中,我们揭示了IAA可以通过AHR上调肠道Papss2的表达。同时,AHR可以结合到Papss2的启动子并启动其转录。Papss2表达的上调增加了细胞内PAPS的生物合成,而Slc35b3的上调促进了PAPS进入高尔基体,这些共同促进了黏液的硫酸化。我们的研究增加了越来越多的数据,表明黏液生化成分的变化可能导致IBD的发病机制,特别是黏液糖链硫酸化修饰。这为基于当前抗炎或其他基于免疫的治疗提供了另一种治疗视角。
往期推荐
