

摘 要
人类肠道微生物群是一个复杂的生态系统,与衰老过程密切相关。然而,目前还没有可靠的方法充分利用肠道微生物群的宏基因组数据来确定宿主的年龄。在这项研究中,我们考虑了地理因素对肠道微生物群的影响,并使用了共计2604个经过筛选的肠道微生物群宏基因组数据来构建一个年龄预测模型。接着,我们开发了一个集成模型,该模型结合了多种异构算法,并结合物种和通路分析进行多视角学习。通过整合肠道微生物群宏基因组数据并调整宿主的混杂因素,模型显示出高准确性(R^2 = 0.599,平均绝对误差 = 8.33年)。此外,我们进一步解释了模型,并识别了衰老过程中的潜在生物标志物。在这些已识别的生物标志物中,我们发现Finegoldia magna、Bifidobacterium dentium和Clostridium clostridioforme在老年人中的丰度增加。此外,随着年龄的增长,肠道微生物群对氨基酸的利用发生了显著变化,这些变化被报告为与年龄相关的营养不良和炎症的风险因素。这个模型将有助于全面利用多种组学数据,并使我们能够更深入地理解微生物与年龄之间的相互作用,实现对衰老的针对性干预。
引 言
在人类中,衰老是一个持续且渐进的过程,导致所有器官系统的生理功能下降。肠道菌群被认为是最重要的共生微生物生态系统,在人类健康中具有多种功能,包括消化、免疫、代谢产物的产生,甚至神经功能。许多队列研究已经表明年龄是影响成年微生物组的主要因素。鉴于预期寿命的快速增加和老年人口比例的上升,加强对肠道菌群与衰老相互作用背后生物学机制的理解是必要的。在这方面,解析肠道微生物组特征对于开发基于微生物组的非侵入性测试以确定老年人加速或延迟衰老的迹象,以及评估基于肠道微生物组的干预措施以缓解与衰老相关的疾病,将是无价的。
宏基因组关联研究已经开始探索衰老过程中的肠道微生物组变化,以研究肠道微生物组与年龄的关联。微生物群的演化并未在儿童时期停止,而是延续到成年甚至老年。近期研究表明,肠道微生物组的α多样性从婴儿期持续增加至成年期,在成年期相对稳定,然后在老年期再次增加。此外,有益细菌(如产生短链脂肪酸的物种)的比例随年龄减少,而机会性物种和病原菌的比例则增加。除了肠道细菌的分类组成外,功能性基因(如与异生物质生物降解和代谢相关的基因)也显示出与年龄的相关性,在老年人中观察到更显著的功能重组。
然而,大多数先前的研究都集中在单一队列上,很少有研究整合了多个人群或其他研究的数据。这些限制阻碍了我们阐明微生物组-年龄关联的稳健性,并模糊了我们对微生物组如何促进衰老的潜在机制的理解。
通过元分析整合所有相关研究的数据,可以评估微生物组-年龄关联的稳健性。研究微生物组在广泛年龄范围内的变化需要一个一致整理的微生物组档案的综合数据集。尽管已经产生了大量关于人类肠道微生物组与年龄关联的宏基因组数据,但从这些数据中获取衰老的分子足迹仍然是一个重大挑战。这些发现是零散的,需要统一到肠道群落动态的理论中。机器学习提供了下一个级别的分析,允许开发与人类肠道微生物组相关的新视角和新假设。人类微生物组老化时钟被认为是衡量肠道群落时间流逝和区分两个时间上不同状态的可靠方法。到目前为止,尽管有大量关于特定微生物与衰老关联的报告,但这一追求在肠道宏基因组学领域产生了有限的结果。口腔、肠道和皮肤微生物组预测年龄的能力以前已经在成人中使用随机森林回归对多个公开可用研究的数据进行组合进行了测试。结果表明,基于皮肤微生物组16s rRNA数据的模型提供了最佳的年龄预测(平均绝对误差[MAE]:3.8年;R^2,0.739)。然而,尽管这些模型在年龄预测方面取得了非常好的结果,扩增子测序在提供微生物组与衰老关联的深入分析方面受到限制。此外,在另一项研究中,提出了一个基于深度神经网络和宏基因组数据的模型(MAE:5.91年;R^2,0.29),但预测的准确性仍然不足,准确性与最终关联解释的可靠性有关。
尽管机器学习算法的强大能力正吸引越来越多的关注,但仍然存在一些限制。首先,微生物组的复杂性及其易受众多协变量和混淆变量(除了年龄以外)的影响,使得将可用信息聚合成一个肠道老化时钟的基本任务变得复杂。其次,由于宏基因组数据通常稀疏,并且以高维度矩阵和高噪声的形式呈现,迫切需要一个最优的机器学习方法来解读衰老与微生物群组成和功能之间的联系。此外,尽管先前的研究已经证明是有洞察力的,但它们对分类学的专注可能限制了我们对微生物组功能与健康之间关联的理解。解决微生物组功能与健康之间关联的能力可能对确定微生物组加速或延迟衰老的机制至关重要。因此,这些不足限制了基于肠道微生物组宏基因组数据建立稳健的年龄回归预测模型的能力,并阻碍了进一步理解肠道微生物组与人类年龄之间潜在相互作用的能力。这些挑战促使引入了一个新兴的机器学习领域——多视角学习,并设想其对肠道微生物群组成和功能的潜在强大应用。特别是,多视角学习比以前用于学习宏基因组数据异质性和揭示交叉谈话模式的综合方法更有效。
本研究的目的是提供一个基于肠道微生物群的功能和组成的通用和可靠的老化时钟。为此,我们使用元分析来纠正多个宏基因组队列之间的数据异质性问题,基于大规模数据集有效地避免宿主或技术因素的影响。此外,我们构建了多视角学习模型,以整合肠道微生物群的分类和代谢途径,旨在建立肠道微生物群与年龄之间的功能和机制解释。最后,我们描述了微生物的丰度和功能如何影响年龄预测,以及如何利用这些知识来识别肠道老化的潜在生物标志物。本研究的结果将提高我们对肠道微生物组与年龄之间关系的理解,为新的老化诊断策略提供见解,并为未来的研究建议潜在的干预目标。
结 果
宿主相关因素对微生物群的影响
共收集了来自31个研究队列的4478个粪便样本,覆盖了13个亚区(图1a,补充表S1),所有样本均来自年龄在18岁以上的个体。采用置换多变量方差分析(PERMANOVA)来分析年龄和其他宿主相关因素对肠道微生物群的分类和代谢途径剖面的影响。地理因素,包括国家和亚区,与肠道微生物群的组成和功能有最大的相互作用,其次是西方化和年龄(图2b)。总的来说,这些结果表明年龄是成年肠道微生物群显著差异的主要因素(经过DNA提取套件和测序平台的调整;Bonferroni校正p < .001)。
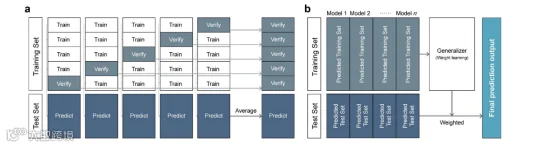
图1. 堆叠集成结构的示意图 (a) 模型集成的第一阶段 (b) 模型集成的第二阶段。
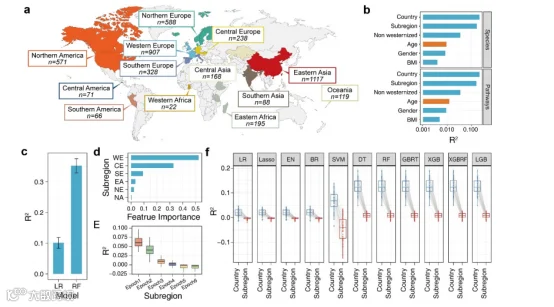
图2. 样本数据概览以及宿主因素与年龄之间的关联。(a) 本研究使用的肠道微生物组宏基因组数据的采样区域(重新分组到亚区级别)和样本大小。(b) 使用Adonis计算宿主因素与微生物组物种和途径组成的效应。(c) 使用线性回归和随机森林模型计算国家和年龄因素的关联。(d) 根据特征重要性得分排序的每个亚区对年龄分布影响的程度。(e) 在每个筛选时期,采样亚区和样本子集年龄因素之间的效应。(f) 使用不同算法计算选定样本子集的国家/亚区和年龄因素之间的效应。以上所有模型的性能通过10次5折交叉验证进行评估。缩写:WE,西欧;CE,中欧;SE,南欧;EA,东亚;NE,北欧;NA,北美。
接下来,我们调查了地理因素分布是否与年龄有关。一般来说,亚区间年龄分布趋势的性质会导致年龄预测模型的性能下降,很难证明与年龄相关的标记不是由地理因素驱动的。此外,鉴于国家和年龄之间的分布特征差异,将地理因素作为额外特征添加到预测模型中,会导致由于特征本身的数据趋势而不是特征与肠道菌群之间的学习关联而产生假阳性情况。出于这些原因,我们使用了两种不同的算法(线性回归[LR]和随机森林[RF])来评估国家和年龄之间的关联。两种算法都显示,在一些国家数据集中确实存在特定年龄范围的样本富集趋势(R2 > 0.1;图2c),这导致了国家和年龄因素之间的错误相关性。
我们结合了两种方法来减少地理位置和年龄之间的相关性,以避免年龄-区域分布问题。首先,我们根据地理位置将国家分组成不同的亚区级别箱子;这确保了在减少特殊分布区域后,仍然保持大多数样本。聚类后,亚区因素仍然是影响肠道微生物群物种和途径的重要因素(图2b)。在亚区级别仍然有一定的相关性(LR, 0.016 ± 0.007; RF, 0.110 ± 0.019)。因此,我们接下来根据聚类结果筛选每个数据集。为了确保统计效力,样本量大于200的亚区级别箱子被考虑用于后续分析。使用RF模型进一步通过特征重要性得分来判断亚区和年龄因素之间的关联强度,依次移除与年龄最相关的亚区箱子(图2d)。最终,我们识别了一个与年龄没有明显相关性的亚区子集(R2 < 0.01;图2e),包括东亚(EA)、北美(NA)、北欧(NE)和南欧(SE)。我们在不同的机器学习算法上评估了这个子集的相关性(图2f)。在所有模型中,筛选后的数据子集有效地避免了与国家级别的分布问题(两者的R2都小于0.01)。
基于分类剖面构建年龄回归模型
为了实现有效的模型集成,基础回归器的多样性是必要的,因为预期异构算法将获得更多的多样性,从而通过集成减少偏差。因此,我们对调整后的数据集(总共2604个样本;数据集中的所有个体年龄在18至107岁之间,中位年龄为52岁)进行了不同机器学习算法的系统评估。
首先,我们的目标是确定哪些模型能够实现肠道微生物群物种级别剖面的年龄预测。总共考虑了11个模型,包括LR、Lasso、Elastic Net(EN)、Bayesian Ridge(BR)、Support Vector Machine(SVM)、Decision Tree(DT)、RF、Gradient Boosted Regression Trees(GBRT)、eXtreme Gradient Boosting(XGB和XGBRF)和LightGBM(LGB)。除了LR和DT模型外,其他几个模型可以实现年龄预测(图3a);实际上,基于树的算法(RF、GBRT、XGB、XGBRF和LGB)比其他模型具有更好的预测性能,而LGB模型的性能最高(R2 = 0.5,MAE = 9.48)。
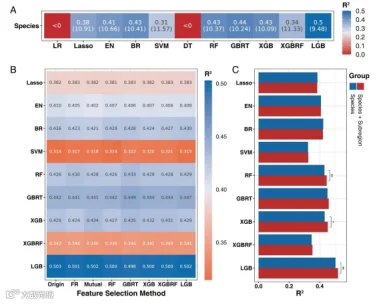
图3. 基于肠道微生物组成预测年龄的模型性能。(a) 不同机器算法预测物种组成年龄的能力(评估指标是R^2和MAE)。(b) 不同特征选择算法对不同模型性能的影响(经过年龄预测能力筛选,评估指标是R^2)。(c) 额外的亚区特征对年龄预测性能的影响(基于特征选择后的物种组成)。
其次,我们采用了一系列的特征选择方法,旨在减少特征维度和计算成本。其中,基于GBRT的选择方法在不降低性能的情况下降低了数据特征维度(图3b,补充表S2)。因此,我们使用GBRT选择的数据作为后续模型集成的输入,考虑到降低了模型计算开销。
此外,我们比较了基于过滤数据的额外亚区标签特征对模型性能的影响(图3c)。基于树的方法(RF、GBRT、XGB、XGBRF和LGB)对额外特征更敏感,结果表明亚区信息可以显著提高模型性能。
基于代谢途径剖面构建年龄回归模型
为了构建一个基于肠道微生物群宏基因组数据的高精度年龄回归模型并提高数据利用率,我们进一步考虑了除了前面提到的分类剖面之外的植物代谢途径的组成。此外,对代谢途径注释结果进行了不同机器学习算法的系统评估。
正如前面提到的,我们首先判断这一系列算法是否能够基于途径数据预测年龄。经过10次5折交叉验证后,除了LR和DT之外的算法显示出不同程度的年龄回归能力(图4a)。有趣的是,与分类剖面相比,途径剖面具有更少的特征维度,但在大多数算法中显示出更好的预测精度(原始物种和途径数据的特征维度分别为904和468)。然而,尽管这两种数据类型在LGB模型中显示出它们的最佳预测能力,但物种和途径之间仍然存在很大的性能差距。总的来说,在这些不同的数据类型中,基于树的算法显示出更好的预测能力,而LGB模型具有最佳性能(R2 = 0.42,MAE = 10.21)。
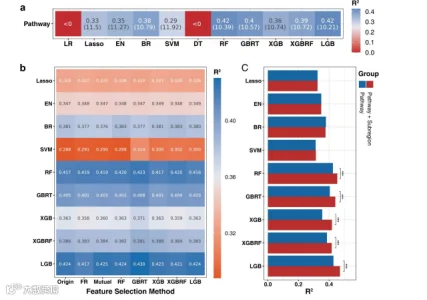
图4. 基于肠道微生物组通路组成的年龄预测模型的性能。(a) 不同机器算法预测通路组成年龄的能力(评估指标是R^2和平均绝对误差MAE)。(b) 不同特征选择算法对不同模型性能的影响(经过年龄预测能力筛选,评估指标是R^2)。(c) 额外的亚区特征对年龄预测性能的影响(基于特征选择后的通路组成)。
接下来,我们实施了相同的特征选择策略,基于GBRT的选择方法在不降低每个模型预测性能的情况下减少了特征维度(图4b,补充表S3)。因此,我们使用GBRT选择的数据进行了后续的模型集成。接着,我们比较了额外的地理特征对基于筛选数据的模型性能的影响(图4c)。与物种建模现象类似,基于树的方法更容易学习亚区特征的影响。相比之下,在途径建模过程中,地理因素显示出更高的性能提升。
基于肠道微生物组宏基因组数据的集成和多视角学习
我们接下来检查了多个模型的融合是否可以提高相比单一模型的预测准确性。为此,我们应用了九种预先验证的回归方法(Lasso、EN、BR、SVM、RF、GBRT、XGB、XGBRF和LGB)来构建集成模型,并使用LR作为泛化器的权重学习算法。首先,我们比较了单一和集成算法在多种数据类型上的预测准确性(分别使用物种集和途径集进行建模,以及使用多个数据集进行建模)。为了评估性能,展示了在两个数据集上准确性最高的LGB作为比较的基准。结果表明,集成方法可以在所有数据类型上显著提高预测准确性,这强调了该方法是可行的(图5a)。此外,多集结果表明,基于多视角的方法可以进一步提高性能。
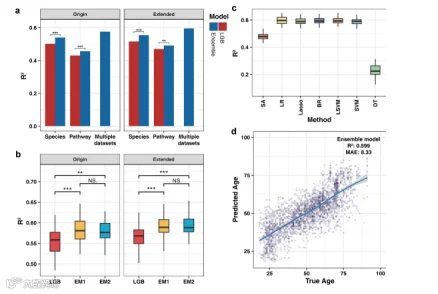
图5. 建模方法的预测准确性。(a) 单一模型和集成模型分析不同数据集的准确性。(b) 不同数据融合方法对模型预测性能的影响。(c) 不同加权方法在扩展数据集上的预测准确性。未标记组之间的预测性能存在显著差异。(d) 集成模型的真实年龄与预测年龄的散点图(基于特征选择的物种和途径数据以及额外的亚区信息)。Origin表示仅经过特征选择的数据集;Extended表示经过特征选择并附加亚区标签的数据集。使用配对Wilcoxon秩和检验分析每组数据之间的差异。
值得注意的是,由于集成算法是对两个数据集进行独立建模,然后将结果作为预测输出进行加权,而大多数当前模型并未设计为同时对多个数据集进行建模,这意味着相应的多个数据集不适合LGB模型。因此,由于数据结构的限制,很难直接比较集成模型和LGB之间的性能差异。为了解决这个问题,我们采用了另一种数据融合方式来比较LGB模型和集成模型之间的预测能力(补充图S1)。为了实现物种和途径数据的综合利用,我们直接连接了两个数据集。每个样本的代谢途径数据与物种数据一起作为额外特征聚合,使用与之前使用亚区特征相同的方法。我们使用连接的数据集构建了LGB和集成模型(简称为EM1),并将它们与使用上述集成策略的集成模型(EM2;先独立训练,再进行结果加权)进行了比较。结果发现,特征的扩展也可以提高回归准确性。在这种集成策略下,我们的集成模型的性能仍然显著高于LGB,且与原始集成方法的性能没有显著差异(图5b)。
我们还测试了不同的权重学习方法,并判断了对集成效应的变化。简单平均(SA)结果被用作不同权重方法的基线,非集成树模型被视为潜在的泛化器,包括LR、Lasso、BR、带线性核的SVM(LSVM)和带非线性核的SVM(SVM)。背后的理念是为了避免复杂模型在泛化过程中的过拟合,从而导致性能损失。我们观察到,基本线性模型(LR、Lasso、BR、LSVM和SVM)能够获得与SA相比理想的集成效果,这些方法之间没有显著差异(图5c)。值得注意的是,DT经历了严重的性能下降,其性能低于基准,这意味着DT可能存在严重的过拟合问题。结合上述结果,我们确定了最简单的LR算法作为集成模型的泛化器,在此基础上,基本学习者可以获得理想的性能。我们的集成模型在所有年龄范围内展示了稳定的预测效果,在任何特定年龄范围内都没有异常的预测偏差(图5d)。
根据集成模型解释肠道微生物组的年龄相关生物标志物
接下来,我们试图解释集成模型,以识别与年龄相关的特征,包括物种和途径两个方面。我们使用置换特征重要性(PFI)方法来研究单个特征与衰老之间的关系。我们获得了一组与衰老过程显著相关的生物标志物(图6a)。其中,肠道微生物组的物种和途径在年龄预测上显示出不同程度的影响。我们观察到,总共有102个微生物物种和41个代谢途径对年龄预测有显著影响(补充表S4)。最具预测性的因素是乙酰辅酶A生物合成、烟酸降解和Finegoldia magna。其余因素包括紫杉烷生物合成、Streptococcus thermophilus、Prevotella copri、己糖发酵、Bifidobacterium dentium和Streptococcus infants。
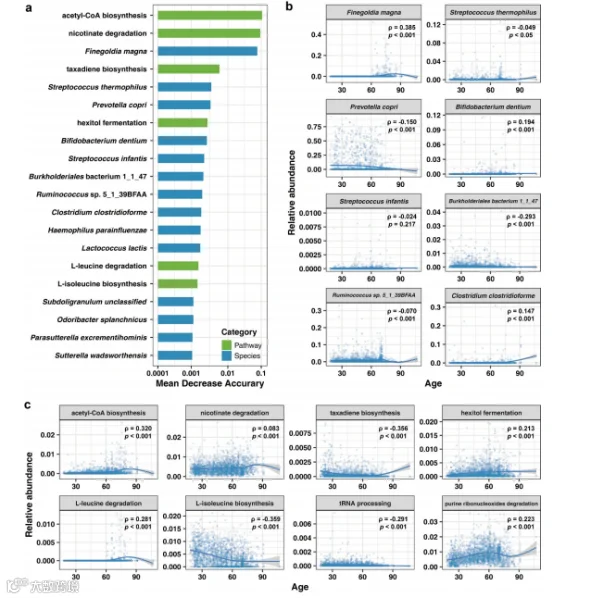
图6. 衰老相关的生物标志物对模型预测性能有显著影响。(a) 在集成模型中对预测年龄模型预测性能影响最大的前20个生物标志物。(b) 受影响最大的前8个微生物物种。(c) 受影响最大的前8个微生物途径。物种/途径与年龄之间的相关性以斯皮尔曼相关系数ρ表示。所有p值都使用Bonferroni校正进行了多重比较调整,并且数据的样条拟合也显示出来(蓝色曲线)。
在这些已识别的生物标志物中,我们发现许多物种和途径随年龄呈现特定趋势。例如,F. magna、B. dentium和Clostridium clostridioforme在老年人口中的丰度增加,而P. copri和Burkholderialse bacterium 1_1_47随年龄减少(图6b)。在代谢途径中也观察到了类似的年龄分布特征;乙酰辅酶A生物合成、烟酸降解和L-亮氨酸降解在肠道中随着年龄的增长而富集的可能性更高。相比之下,紫杉烷生物合成、tRNA处理和L-异亮氨酸生物合成随年龄增长而功能丧失的可能性很高(图6c)。
讨 论
机器学习算法的发展为肠道微生物组数据的全面和深入分析提供了新的机会,并使我们能够将微生物复杂的物种和途径组成与宿主状态相关联。在本研究中,我们构建并评估了一个新颖的集成建模框架,使用超过2500个肠道微生物组的宏基因组测序数据进行了通用的年龄回归。我们的集成模型比目前使用的方法实现了更好的预测准确性和更高的数据利用能力。值得注意的是,我们的研究独特之处在于,我们不仅纠正了地理位置因素对宿主菌群的影响,而且还在肠道微生物组研究领域整合了异构算法和多视角学习。
基于肠道微生物组精确构建一个老化时钟对于解释肠道菌群在衰老过程中的作用至关重要,因为它可以指导随后的抗衰弱干预措施。宿主背景信息的多样性使得每个样本的肠道微生物组都是独一无二的,这将影响基于肠道菌群的诊断准确性。事实上,在某些情况下,这种个体变异甚至可能掩盖微生物组与宿主状态之间的实际关系。因此,应该避免由于背景因素的分布特征而导致的无效相关性,以构建准确的年龄预测模型。先前的研究表明,纠正混杂因素可以改善肠道微生物组变化的识别和解释效果。在各种因素中,地理因素被认为是影响肠道微生物组结构的主要混杂因素。因此,我们进行了一项全面的元分析,重点关注整个年龄范围(除未成年人外),以扩展对人类衰老的更广泛景观的发现;这可能导致发现更普遍的与年龄相关的微生物组变化模式。同时,我们应用了两步筛选方法来实现样本年龄和国家的解耦。我们的结果显示,上述方法可以解耦地理和年龄因素。更重要的是,通过在肠道微生物组物种或途径组成中添加亚区的额外特征而实现的性能提升,并不是由亚区和年龄之间的直接关系引起的,而是通过学习不同地理区域的独特菌群特征。
使用现代高通量测序技术可以对整个微生物群落的全基因组进行分析;然而,相应的菌群序列数据集的大小和复杂性也增加了,如何有效管理、分析和整合这些高维度大数据已成为一个重大挑战。最近,机器学习被用来解决这些问题,因为它能够实现考虑菌群之间相互作用的解释,增加我们对现有数据结构的理解。大量的微生物组研究已经应用机器学习方法建立了疾病诊断模型,并探索了潜在的关联,如癌症、心血管疾病和糖尿病。因此,在解构区域因素的影响后,我们对多种异构模型算法进行了系统比较,其中每个回归器在宏基因组数据分析中都被广泛使用。由于算法机制的不同,每个回归器可能对相同的输入数据集有不同的输出。然而,通过利用模型的多样性,可以构建一个更准确、偏见更少的回归模型。因此,确定能够实现年龄预测的基础回归器对于实现模型集成至关重要。除了考虑模型算法,我们还进一步构建了基于肠道微生物组代谢功能的预测模型。已经提出,微生物功能可能比分类组成更具信息性和保守性。对这两种数据类型的建模结果表明,大多数模型可以实现基于肠道微生物组宏基因组数据的年龄预测,途径在大多数模型中表现出更好的结果。此外,还进行了额外的测试,以比较对亚区因素的修正对预测准确性的影响,结果表明,通过考虑宿主混杂因素可以进一步提高预测精度。总之,我们的综合分析揭示了物种和途径都与人类衰老过程相关,调整地理位置因素可以改善对衰老中肠道微生物组变化的识别。
为了提高准确性并实现多组数据的综合利用,我们使用了堆叠策略来构建集成模型。集成方法已被广泛用于增强模型的预测能力,因为它们既有效又易于实施。尽管集成学习已逐渐应用于基于基因表达的癌症诊断等生物分析中,但在当前的微生物学研究中,它仍然局限于单个模型。然而,与建模过程中的单一算法相比,集成算法的相应成本导致了数据计算开销的大幅增加。为了减少模型构建的运行时间,使用了不同的特征选择方法来减少特征维度。特征过滤后,所选数据集显示出减少的计算时间和略微提高的模型性能,从而在性能和速度之间实现平衡。所呈现的实验强烈证实了整合方法在三个数据集(包括物种、途径和两组的组合)上的有效性,并表明多模型集成的准确性高于所有数据集上单独行动的任何单个回归器。然而,以前的研究倾向于关注物种或基因来分析微生物与宿主状态之间的关系;因此,包含大量关于肠道微生物组信息的宏基因组测序数据未能得到充分利用。在本研究中,我们不仅限制于集成异构模型,还同时整合了不同类型的数据,这使我们能够实现多视角建模。我们的结果表明,物种和途径的结合为模型提供了更好的预测能力,并证实了以不同方式综合考虑注释数据可以描述宿主肠道微生物组的整体状态。我们解决了以前研究中解释潜力弱(扩增子测序的低分类注释水平)和预测准确性低的限制。我们还评估了不同的数据融合策略,以证明这些方法之间没有性能区别。在集成之前对每个数据集进行建模的优势在于,与首先合并数据集相比,解耦数据集可以提高数据利用率;事实上,当多个数据集之间的样本不完全匹配时,独立建模具有更好的适应性。然而,首先连接的策略无法处理由样本不一致性引起的大量缺失值,例如样本仅提供物种组成而缺乏功能注释,或者样本已经进行了全基因组测序,但没有测量代谢组数据。因此,这些样本只能直接丢弃,最终导致数据的浪费。首先建立模型的策略可以通过利用所有数据来构建模型来有效解决这个问题。在这种策略中,只需要部分匹配数据进行最终泛化器训练,以结合分别训练的模型,最大限度地利用数据集。尽管本文没有涵盖这种情况,但高数据利用率是必要的,尽管肠道菌群的组学技术不断发展并为相关研究提供数据支持,但同一样本的多组学数据,如宏基因组学、转录组学和代谢组学,仍然非常稀缺。此外,大多数以前的多组学研究分别揭示了每个数据集中的模式;因此,可能很难检测到未被单一组学数据类型挖掘暴露的一些微调结构。我们的计算框架可以实现多组学数据的有效利用,以分析肠道微生物组与宿主状态之间的全面关联,并可能帮助阐明跨组学的复杂机制。
通过依赖更准确的年龄预测模型,我们使用广泛的年龄范围的全面宏基因组注释数据揭示了潜在的衰老生物标志物。总共有102个物种和41个途径被认为与衰老过程密切相关。值得注意的是,许多生物标志物与衰老相关疾病或在老年人中更普遍的疾病相关。例如,克雷伯菌是革兰氏阴性菌血症感染的主要原因之一,老年患者有更高的感染风险。此外,F. magna和P. copri与关节炎相关,与此一致,与醋酸生产相关的途径(与关节炎相关)也被识别为生物标志物。与以前的一项研究一致,我们识别出的物种和途径中有相当一部分与老年人口的虚弱有关,包括C. clostridioforme、Clostridium hathewayi、Clostridium bolteae、Clostridium leptum、Clostridiales bacterium 1_7_47FAA和丙酮酸发酵途径。此外,一些指标可能阐明肠道环境的集体特征。在先前获得的生物标志物中,我们观察到肠道微生物组对氨基酸的利用随年龄增长而发生显著变化。这种导致氨基酸减少的代谢特征可能加剧衰老过程中的营养不良,这不利于维持免疫系统功能和预防虚弱。值得注意的是,随着年龄的增长,支链氨基酸(BCAAs)的消耗增加,尤其是亮氨酸代谢的富集和异亮氨酸合成途径的丢失。BCAAs被认为可以改善肌肉蛋白质合成,相关研究表明,老年人口需要消耗更高水平的亮氨酸;这些发现与我们分析揭示的关键代谢特征一致。此外,与色氨酸生物合成相关的途径也被认为与衰老有关。犬尿氨酸途径被认为是人类色氨酸代谢的主要途径。犬尿氨酸具有神经毒性,可以直接损害线粒体,从而导致与衰老相关的炎症。肠道菌群中色氨酸的合成可能导致体内犬尿氨酸的积累。这些与年龄相关的变化最终可能导致疾病。这些与年龄相关的生物标志物阐明了肠道微生物组在衰老过程中的潜在作用,集成方法使我们能够对不同类型的数据进行排序,以实现对衰老的更精细的方向控制。
当前研究包括一个多视角数据适应的集成机器学习框架。通过异构模型和数据的整合,我们观察到了衰老过程中肠道微生物组变化的广泛模式,并澄清了物种和功能在这一过程中的影响。未来的研究应该集中在进一步改进肠道微生物组大数据的收集上。虽然我们在本研究中去除了一些宿主混杂因素,但需要更多高质量和多样化的数据来探索肠道细菌与年龄之间的深入关联。机器学习框架也应该得到改进。事实上,不断有新算法被提出,特别是神经网络模型,在其他领域显示出相当的预测潜力。通过方法的改进,将可能扩展数据以确保结论的可靠性并最大化其效用。
往期推荐

