
奇辉观点
近年的研究显示肠道微生物组在预测性医疗应用中发挥了关键作用。在本研究中作者介绍了肠道微生物群健康指数2(Gut Microbiome Wellness Index 2,简称GMWI2),这是基于肠道微生物组分类学档案的标准化健康状态指标,用于预测健康和疾病状态。研究涉及汇总来自全球人口景观(涵盖26个国家和六大洲)的54项已发表研究中的8069个粪便宏基因组数据,以识别与疾病存在或缺失相关的肠道分类信号。GMWI2采用了Lasso惩罚逻辑回归模型,在区分健康(无疾病)与非健康(患病)个体方面的交叉验证平衡准确率达到80%。这一表现超过了原始GMWI模型和传统的物种水平α多样性指数,表明了一个更强大的肠道微生物组特征,用于区分多种疾病中的健康和非健康表型。通过跨研究验证和外部验证队列进行评估时,GMWI2保持了近75%的平均准确率。此外,通过重新评估先前发表的数据集,GMWI2为饮食、抗生素暴露和粪便微生物群移植对肠道健康的影响提供了新的见解。作为一个开源命令行工具,GMWI2代表了使用个体独特的肠道微生物组成来评估健康的及时而关键的资源。虽然GMWI2不用于确诊具体疾病,但它可以作为一种早期警告系统,提示可能的健康变化;同时,GMWI2的开发鼓励了进一步研究肠道微生物组与复杂慢性疾病之间的联系,并探索如何利用这些微生物特征来改善健康。这项研究通过开发GMWI2这一新的健康状态预测工具,为理解肠道微生物组与人类健康之间的关系提供了新的视角,并为未来的个性化医疗和公共卫生干预提供了潜在的工具。
论文ID
本文译自:Chang D, Gupta VK, Hur B, et al. Gut Microbiome Wellness Index 2 enhances
health status prediction from gut microbiome taxonomic profiles. Nat Commun. 2024 Aug 28;15(1):7447. doi: 10.1038/s41467-024-51651-9.
发表杂志:Nat Commun
杂志影响因子:16.1
通讯作者:Jaeyun Sung
作者单位:Mayo Clinic, Rochester, USA
正文
近期具有里程碑意义的研究表明,肠道微生物组与多种复杂慢性疾病之间存在深刻的联系。尽管有这些发现,我们如何判断一个人是否患有菌群失调?我们如何有效地利用独特的微生物特征来定量追踪我们的健康?这些关键问题站在利用肠道微生物组作为健康和福祉精确标志的前沿。
肠道微生物组作为解码复杂慢性疾病的潜在标志物,已经吸引了科学界的极大兴趣--作为回应,我们最近开发了肠道微生物群健康指数(Gut Microbiome Wellness Index,简称GMWI)。GMWI是一种首创的基于粪便宏基因组的指标,通过确定个体仅从其肠道微生物组成中携带临床诊断疾病的可能性来评估健康,而不考虑特定疾病类型。这一与疾病无关的指数是从包含来自34项独立研究的4347个粪便宏基因组数据的综合分析中得出的。GMWI是健康和疾病相关肠道微生物物种的集体丰度的对数比率--一个包含物种水平相对丰度和多个α多样性指标的术语。在汇总数据集上评估时,GMWI在预测临床诊断疾病存在时的平衡准确率(即正确分类的健康和非健康样本比例的平均值)为69.7%。具体来说,健康(无疾病)个体和非健康(患病)条件的正确分类率分别为75.6%和63.8%。此外,GMWI在679个粪便宏基因组的验证队列中实现了73.7%的平衡准确率,健康和非健康子集的正确分类率分别为77.1%(118个中的91个)和70.2%(561个中的394个)。自2020年首次发表以来,GMWI已被用于研究环境和遗传/社会经济因素对人类肠道微生物组的影响,以及在识别‘长寿肠道微生物组特征’物种集群的研究中。
尽管我们最初的GMWI原型充满希望,但仍有一些限制阻碍了其普遍适用性。首先,GMWI在正确分类健康粪便宏基因组方面的成功率高于非健康样本。这种偏差可能源于用于识别与健康相关和与疾病相关物种的基于流行率的策略,这是GMWI模型的一个基本组成部分。由于非健康组包括患有不同疾病患者,这一组本身是异质的;反过来,基于流行率的策略可能会错过仅在非健康人群的子集(例如,具有特定疾病的队列)中表现的微妙分类特征。其次,我们现有的模型为每个物种分配了相同的权重,而没有考虑个别物种重要性可能存在的变异。为了提高分类准确性和普遍适用性,需要一个精细化的权重系统,该系统考虑与宿主表型关联的不同强度。此外,包括来自所有分类等级的肠道微生物信息可能会发现更多能够准确预测宿主表型的特征。在这项研究中,我们介绍了GMWI2,这是原始GMWI的一个高级迭代版本,它解决了上述限制,并显著提高了在区分健康和非健康表型方面的分类准确性。
结果
正如我们之前的工作中定义的,“健康”受试者是指那些没有报告疾病或异常体重状况的人(即,根据报告的BMI被分类为体重不足、超重或肥胖),而“非健康”受试者是指那些被确认患有任何疾病的临床诊断的人。(保留“健康”和“非健康”的相同定义,确保当前工作代表了我们原始GMWI方法的持续改进。)我们对现有的8069个粪便宏基因组进行了汇总分析(其中5547个来自健康个体,2522个来自非健康个体),这些数据来自54项独立发表的研究,涵盖了26个国家和六大洲(图1a,表1和补充数据1)。这些汇总的宏基因组来自具有十二种不同健康和疾病表型的个体(图1a;健康、强直性脊柱炎、动脉粥样硬化性心血管疾病、结直肠癌、克罗恩病、格雷夫斯病、肝硬化、多发性硬化症、非酒精性脂肪肝病(也称为代谢功能障碍相关脂肪肝病[MASLD])、类风湿性关节炎、2型糖尿病和溃疡性结肠炎),来自不同的地理位置、种族/人种、文化,并平衡了性别代表性(图1b)。(我们的研究和样本选择标准可以在“方法”部分找到。我们在补充数据2中提供了所有受试者的表型、年龄、性别、BMI和地理信息[如他们各自原始研究所提供的]。)样本量的显著增加,几乎是我们之前研究中包含的宏基因组数量的两倍,是GMWI2的一个显著改进。此外,GMWI2使用MetaPhlAn316而不是MetaPhlAn217进行分类分析,利用一个大大扩展的标记数据库,以更全面和准确地表征微生物分类群(“方法”部分)。

图1 | 对来自全球多样化代表的多种健康和疾病状况的粪便宏基因组进行汇总分析。
所有的宏基因组都经过了统一的重新处理,使用了相同的生物信息学流程,如“方法”部分所述。这种做法不仅减轻了批次效应,而且还加强了在可能存在强烈混杂因素的情况下识别与健康和疾病相关的肠道分类特征的能力。实际上,这得到了主成分分析(PCA)的支持,尽管样本来自不同的来源和条件,健康和非健康组显示出显著不同的肠道微生物组轮廓(Adonis R2 = 1.2%, P = 0.001, PERMANOVA;图2a)。尽管如此,尽管对宏基因组数据的共识预处理有效地减少了与生物信息学分析相关的批次效应的一个来源,但重要的是要认识到这种方法不能完全消除不同研究中实验和技术程序引起的潜在批次效应。这些因素包括粪便样本的收集、储存和准备进行宏基因组测序的方式的差异。
在GMWI2中实施Lasso惩罚逻辑回归
对于区分健康和非健康组的分类任务,GMWI2使用Lasso惩罚逻辑回归模型,而不是原始GMWI中使用的对数比率方程。因此,GMWI2本质上使用线性回归进行预测,类似于统计遗传学中的多基因风险评分模型。该模型在所有可测量的分类等级上训练,以模拟疾病可能性作为微生物分类群(即,类群)存在或缺失的线性函数。具体来说,GMWI2分数为个体样本定义为样本来自健康、无疾病个体的预测对数几率(logit)。关于GMWI2如何使用Lasso惩罚逻辑回归来估计疾病可能性的更全面解释,详见“方法”部分。
原始的GMWI方法采用了基于流行率的策略来识别与健康和疾病相关的微生物物种。我们当前的方法学习了变量特征的重要性,消除了手动物种识别的需要。更具体地说,所使用的Lasso惩罚逻辑回归模型使用了95个具有非零系数的微生物分类群来进行预测,这些分类群直接从肠道微生物组档案中得出(图2b和补充数据3)。有趣的是,大多数具有正系数和负系数的分类群在健康和非健康组中的相对丰度较高(补充数据4)。这些被识别的分类群包括1个类、3个目、4个科、19个属和68个种。值得注意的是,系数值在-0.68到0.54之间变化,确保每个分类群根据其相对关联强度的不同对GMWI2分数做出不同的贡献。这与我们之前的GMWI对数比率模型不同,后者为每个物种分配了相等的权重。
值得一提的是,在我们的分析中,几个分类等级展示了非零系数。这部分可能是因为不同分类等级层次之间的相互依赖引入了多重共线性,这使得回归系数的解释变得复杂。然而,我们包含所有分类等级的方法在分类性能上比仅使用单一分类等级时表现出更高的性能(补充表1)。鉴于我们的主要目标是优化分类准确性,我们选择优先考虑这一点,从而将多重共线性的问题放在一边。
在接下来的部分中,我们评估GMWI2在区分健康与非健康个体方面的熟练程度。这个过程可以概念性地分为四个阶段:
1. 模型训练:GMWI2在完整的训练数据集上进行训练和评估。这个阶段使用所有8069个样本来计算逻辑回归系数(如图2b所示)并确定GMWI2分数。
2. 交叉验证:GMWI2通过交叉验证(CV)和跨研究验证(ISV)策略进行进一步评估。与初始阶段不同,这些策略不会同时使用所有8069个样本进行模型训练。因此,这个阶段生成的模型与第一阶段产生的模型本质上是不同的。按照标准的交叉验证协议,GMWI2模型的训练,包括逻辑回归系数的计算,严格限制在每次训练-测试分割的总8069个样本的训练部分。
3. 在外部数据集上的验证:在第一阶段开发的GMWI2模型应用于六个外部数据集,以确认其在独立样本上的鉴别能力。
4. 在纵向数据集上的演示:第一阶段的GMWI2模型应用于四个额外的外部数据集。这些评估专注于展示GMWI2在纵向场景中的适用性。
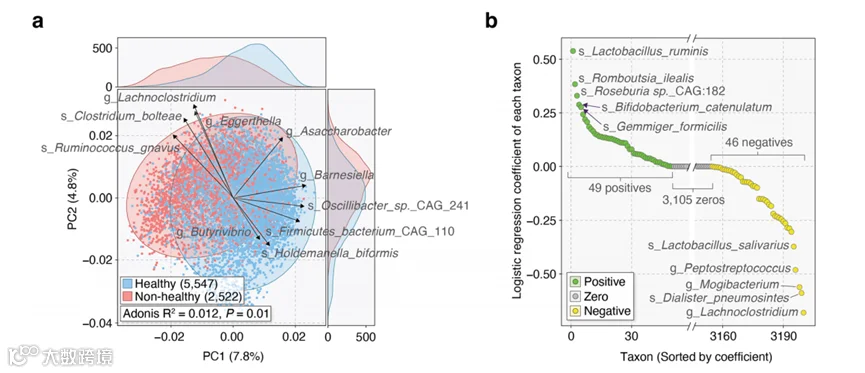
图2 | 健康和非健康个体的肠道微生物组分类档案构成了一个Lasso惩罚逻辑回归分类模型。
使用GMWI2增强健康和非健康肠道微生物组的分类
GMWI2分数是通过应用学习到的系数来计算预测的对数几率得出的。正值的GMWI2将样本分类为健康,表示没有疾病;而负值的GMWI2将样本分类为非健康,表示疾病存在。GMWI2为0意味着正系数分类群和负系数分类群的加权存在相等,因此将样本分类为既不是健康也不是非健康。
在训练数据集(8069个样本)上评估时,GMWI2展示了79.9%的平衡准确率(健康的正确分类率:79.2%,非健康的正确分类率:80.6%)和Cliff's Delta(d)效应大小为0.75,显著超过了我们原始GMWI模型(71.8%,d = 0.63)和传统物种水平α多样性指数(即Shannon指数、Simpson指数和丰富度)报告的平衡准确率和Cliff's Delta(图3a和补充数据5)。我们的结果表明,GMWI2比GMWI更有效地区分健康和非健康组,尽管两个指数有很强的相关性(Pearson's r = 0.81;补充图1)。此外,我们发现健康个体的肠道微生物组与十一种疾病表型相比,GMWI2分数显著更高(图3b)。最后,我们观察到GMWI2与临床/人口统计特征之间的相关性较弱(|Spearman's ρ| < 0.3;补充图2a-g),如年龄、BMI、空腹血糖、血脂和甘油三酯,表明这些因素不会显著影响基于肠道微生物组的分类结果。
我们随后探索了更高的(或更积极的)GMWI2值是否能够表示对将粪便宏基因组归类为健康的信心增强。相反,我们检查了更低的(或更消极的)GMWI2分数是否表明一个样本被归类为非健康的可能性增加。事实上,我们观察到,随着GMWI2分数越来越积极,健康个体在宏基因组样本中的比例逐渐增加(图3c和补充表2)。同样,越来越消极的GMWI2分数捕捉到了更大比例的非健康受试者。值得注意的是,与GMWI的正负箱中的实际健康和非健康样本比例相比,GMWI2的正负箱中的比例都更高(参见图3c中的点)。GMWI2和GMWI箱中样本分布的这种差异突显了GMWI2在区分健康和非健康样本方面的改进能力。
我们研究中图3c所示的结果揭示了一个有趣的趋势。具体来说,当GMWI2(和GMWI)分数表现出更积极或消极的值时,实际健康和非健康样本的比例相应增加。这一趋势表明,表型分类的信心可能增加。相反,当这些值接近零时,我们准确判断疾病存在或不存在的信心就会下降。为了更仔细地检查这一点,我们接下来研究了设置最低GMWI2阈值或截止参数如何能够提高表型预测的分类准确性。我们观察到,在考虑GMWI2分数幅度增加的截止值时,分类性能有了显著提高,从而意味着对保留样本的预测信心更高(补充表3)。例如,当保留GMWI2幅度等于或高于0.5(即,GMWI2分数低于-0.5或高于+0.5)和1.0(即,GMWI2分数低于-1.0或高于+1.0)的样本时,我们分别实现了85.8%和91.0%的平衡准确率(图3d)。(这些截止值是说明GMWI2幅度截止概念的例子。)然而,这种方法需要排除低于这些截止值的GMWI2幅度的样本,分别只剩下6364(占总数8069样本的78.9%)和4712(占8069的58.4%)个样本。这突出了一个重大的权衡:提高截止值可以提高准确性,但会从分析中排除可能有价值的样本。
一个重要的观察是,尽管样本数量不平衡,GMWI2仍然以几乎相同的比率正确分类了健康和非健康的粪便宏基因组(分别为79.2%和80.6%)。这与原始GMWI形成鲜明对比,后者在健康样本上实现了更高的正确分类率(图3e)。我们还评估了使用留一法交叉验证(LOOCV)和10折交叉验证(10-fold CV)的GMWI2模型的性能(图3e)。有趣的是,GMWI2在LOOCV和10折CV中分别实现了几乎相同的平衡准确率79.1%(健康正确分类率:78.6%,非健康正确分类率:79.5%)和79.0%(健康正确分类率:78.6%,非健康正确分类率:79.3%),几乎与训练数据集上实现的性能(79.9%)相匹配。
接下来,我们使用两种交叉验证方法的不同幅度截止值计算分类准确率(图3e)。值得注意的是,GMWI2在LOOCV和10折CV中分别在分数低于-1.0或高于+1.0的样本上实现了90.4%和90.2%的平衡准确率。这些平衡准确率非常接近在训练集上观察到的(91.0%)。相比之下,当将相同的标准应用于GMWI(即,截止值为1.0)时,平衡准确率显著下降至78.6%。总的来说,这些结果强调了GMWI2相比GMWI所取得的显著改进。
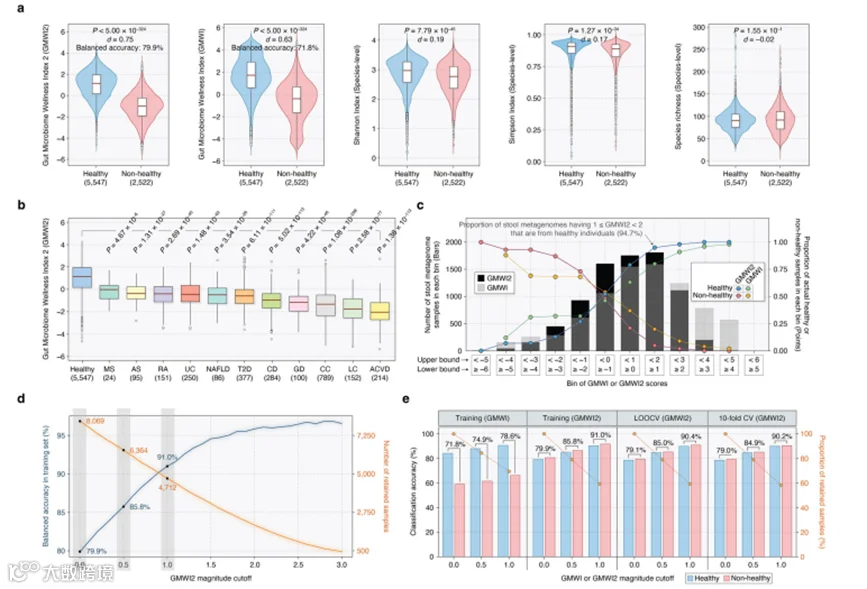
图3 | 使用肠道微生物群健康指数2(GMWI2)增强健康和非健康粪便宏基因组的分类。
评估GMWI2在不同样本大小的研究人群中的稳健性
尽管小样本量的研究被排除在训练集之外(见图1a中的研究排除标准和“方法”部分),但通常,对不同样本大小的数据集验证任何分类模型的稳健性至关重要。为此,我们进行了跨研究验证(ISV),以评估批次效应(即,与研究人群或地点特征相关的技术或生物学变化)对GMWI2性能稳定性的影响。在这种方法中,我们迭代地排除一个研究,对其余研究进行GMWI2模型的训练,并在保留的研究上评估其分类性能。(被排除的研究本质上成为了独立的验证[或测试]队列。)ISV的一个重要方面是,它可以展示出根据验证集的选择可能出现的显著的分类性能变化。对于我们的研究,它提供了在54个独立验证集上应用GMWI2时可实现的分类准确率范围。
图4a特别展示了GMWI2在所有保留研究中的性能,以及它们的样本大小的详细信息。尽管不同研究之间的分类性能存在变化(见图4a中指示每项研究ISV分类准确性的金色点和补充表4),平均平衡准确率为75.8%。当考虑GMWI2分数低于-1或高于1的样本时,这一性能上升到了86.9%(补充表4)。总的来说,我们的分析没有发现模型的预测性能与保留数据集的样本大小之间存在明显的相关性。
从ISV获得的分类性能与LOOCV和10折CV获得的性能相比,差异很小,后两者不考虑研究边界。这些策略之间的小差异显示了GMWI2对批次相关偏见的抵抗力,表明GMWI2能够有效地泛化到粪便宏基因组上,无论受试者的来源如何。这种稳健性的进一步证据是通过训练集、10折CV和ISV中的曲线下面积(AUC)指标展示的,分别实现了0.88、0.87和0.84的AUC(图4b)。
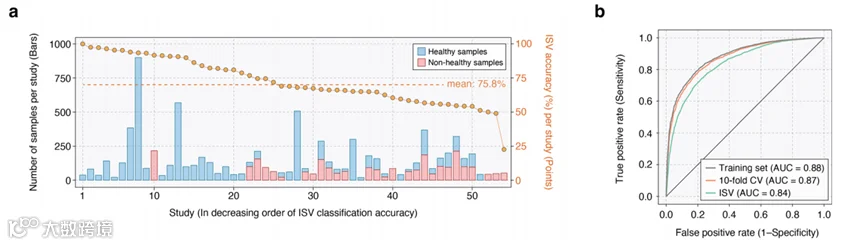
图4 | 跨研究验证(ISV)显示GMWI2在不同研究人群中的有效泛化。
展示GMWI2在独立样本集上的预测能力
为了确认GMWI2在区分健康和非健康个体方面的预测能力,我们汇编了一个包含来自六项已发表研究的1140个粪便宏基因组样本的外部验证数据集(补充数据6)。这个数据集包括健康个体和被诊断为强直性脊柱炎、胰腺癌或帕金森病的患者的样本。这个验证数据集中的所有宏基因组样本(补充数据7)都按照上面展示的方式被分类为健康或非健康组。
与我们在发现队列(或训练数据)中的发现一致,健康验证组(n = 494)的粪便宏基因组的GMWI2分数明显高于非健康验证组(n = 646)的分数(P = 1.6 × 10^–43,双侧Mann–Whitney U检验;Cliff’s Delta = 0.48;图5a)。实现的平衡准确率为72.1%,与我们在ISV分析中观察到的平均75.8%的平衡准确率相当。当幅度截止值为0.5和1.0时,平衡准确率分别提高到75.4%和80.1%,同时仍然保留了74.3%和49.3%的样本。
为了进一步检查GMWI2在外部验证数据上的性能,我们分析了八个总的队列(由每项研究的唯一表型定义),涵盖五个健康和三个非健康表型。如图5b所示,五个健康队列中的四个(H1–H4)的GMWI2分布明显高于所有三个非健康表型队列(P < 0.01,双侧Mann–Whitney U检验)。五个健康队列的分类准确率如下:H1为96.3%(135中的130),H2为91.2%(57中的52),H3为83.3%(30中的25),H4为56.8%(37中的21),H5为28.1%(235中的66)。或者,三个非健康队列的分类准确率如下:胰腺癌(PC5)为90.7%(43中的39),帕金森病(PD6)为81.2%(490中的398),强直性脊柱炎(AS4)为80.5%(113中的91)。值得注意的是,尽管帕金森病患者的粪便宏基因组并未包含在原始发现集中,但GMWI2在预测帕金森病的不良健康状况方面表现良好(81.2%)。此外,尽管H5队列的分类性能相对较差(28.1%),但H5中的GMWI2分数明显高于同一研究中的PC5胰腺癌组。总体而言,GMWI2在外部验证数据集上的稳健可重复性表明,在数据集整合和指数制定过程中,有效地捕获了跨多种疾病的肠道微生物群失调的一般化疾病相关特征。
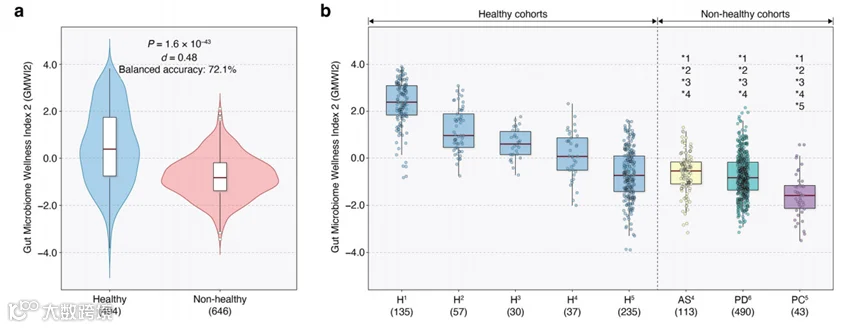
图5 | GMWI2在健康和非健康外部验证队列上的表现。
在纵向研究中追踪肠道健康
我们将GMWI2应用于最近发表的四项纵向肠道微生物组研究中获得的粪便宏基因组。重要的是,这些样本不属于用于训练GMWI2的初始8069个宏基因组的池。在这里,我们的目标是通过展示GMWI2在肠道微生物组健康追踪方面的多功能性,来说明其适用性超出了最初预期的病例与对照场景。我们用于定量监测肠道健康的指数可以类比于使用胆固醇和葡萄糖测试来评估随时间变化的心血管和代谢健康。
利用第一项研究的数据,我们分析了22名肠易激综合症(IBS)患者在接受两名健康捐赠者的粪便微生物群移植(FMT)前后的粪便宏基因组。在参与者中,有14人在FMT后报告症状缓解(“效果”组),而8人尽管在FMT后的六个月内物种丰富度显著增加(P < 0.05,单侧Wilcoxon符号秩检验;补充图3),但没有经历症状缓解(“无效果”组)。然而,只有“效果”组的个体显示出GMWI2的显著增加(P < 0.05;图6a和补充表5)。同样,只有在“效果”组中观察到物种水平Shannon指数的增加(P < 0.05;补充图4)。
总体而言,这些发现表明,虽然α多样性指标,如丰富度和Shannon多样性,可能会得出相互矛盾的结论,但GMWI2的变化可以作为FMT治疗IBS后受试者表型的标记。此外,鉴于FMT供体筛选的临床意义和所涉及的复杂性,像GMWI2这样的计算工具(鉴于其对肠道健康更细致的定义)可能有助于指导选择合适的健康捐赠者及其粪便样本。
在第二项研究中,我们调查了饮食的影响。我们计算了30名健康志愿者在饮食干预前后获得的粪便宏基因组的GMWI2。研究了三组参与者:素食者(自报为素食者并恢复了他们的常规饮食)、杂食者(参与者食用了动植物来源的标准饮食)和专一肠内营养(EEN)(在研究期间转为食用合成的无纤维饮食的杂食者)。在基线时和饮食干预期间的每一天都收集了粪便样本。我们观察到,素食者和杂食者的GMWI2分数在整个五到六天的干预期间保持相对稳定(图6b)。然而,EEN组的GMWI2从第二天起就显著低于基线(P < 0.05,双侧Wilcoxon符号秩检验;图6b和补充表6),而α-多样性在各组间没有显著变化(补充图5)。这些结果表明,去除膳食纤维可能导致整体肠道健康的快速下降,这是GMWI2而不是α-多样性指标唯一检测到的早期变化。总体而言,我们的发现加强了膳食纤维对健康益处的证据。
在第三项研究中,我们为12名健康年轻成年人在接受了4天广谱抗生素(美罗培南、庆大霉素和万古霉素)暴露后的粪便宏基因组计算了GMWI2。在这里,粪便样本在暴露前收集,然后在干预后的第4天、第8天、第42天和第180天再次收集。虽然物种水平的α-多样性测量(Shannon指数和丰富度)表明肠道微生物组可能在第42天或第180天有所恢复,但GMWI2即使在第180天也没有显示出任何恢复趋势(图6c和补充表7)。这些发现反映了Palleja等人最初注意到的干预后有害的分类变化,例如之前无法检测到的梭菌属的增加,以及益生菌双歧杆菌和丁酸产生菌Coprococcus eutactus和Eubacterium ventriosum的消失。因此,我们的结果为短期广谱抗生素干预对肠道微生物群落的长期影响提供了一个新的视角,并表明GMWI2可能是评估急性疾病后肠道微生物组恢复的有价值工具。
在最后一项研究中,我们检查了各种低聚糖对肠道微生物群落的影响。在这项研究中,Lee等人使用GMWI评估了低聚糖的益生元效应,这对设计基于它们对肠道微生物组健康影响的个性化饮食具有更广泛的意义。在此,19名健康成年志愿者(14名男性和5名女性)提供了粪便样本,然后将这些样本混合并充分搅拌。接着,将低聚果糖(FOS)、低聚半乳糖(GOS)、木寡糖(XOS)、菊粉(IN)和2'-岩藻糖乳糖(2FL)分别与同质化粪便样本的一部分混合,在24小时厌氧批量粪便发酵系统中进行。还包括两个对照组:一个在0小时没有添加底物的(NS0)和另一个在24小时没有添加底物的(NS24)。每个研究组进行了三次实验。
对所有粪便样本计算了GMWI2(图6d和补充表8),从而使用我们的新指数复制了原始研究。与之前的发现一致,NS24组的平均GMWI2低于NS0组,表明其健康状况较差,与疾病相关的程度更高。值得注意的是,与NS0相比,添加三种益生元(FOS、IN和GOS)的GMWI2显著更高(P < 0.05,Tukey的HSD检验)。同样,这三种益生元以及XOS与NS24相比,GMWI2也显著更高(P < 0.05)。然而,与GMWI2结果不同,传统的α-多样性指标(Shannon指数、物种丰富度、物种均匀度和逆Simpson指数)在所有益生元处理组中与NS0组相比显著降低(P < 0.05)。因此,至少在体外发酵环境中,这四种益生元的摄入可能会刺激与健康条件相关的肠道微生物物种的生长,这种效应仅通过使用GMWI2观察到。
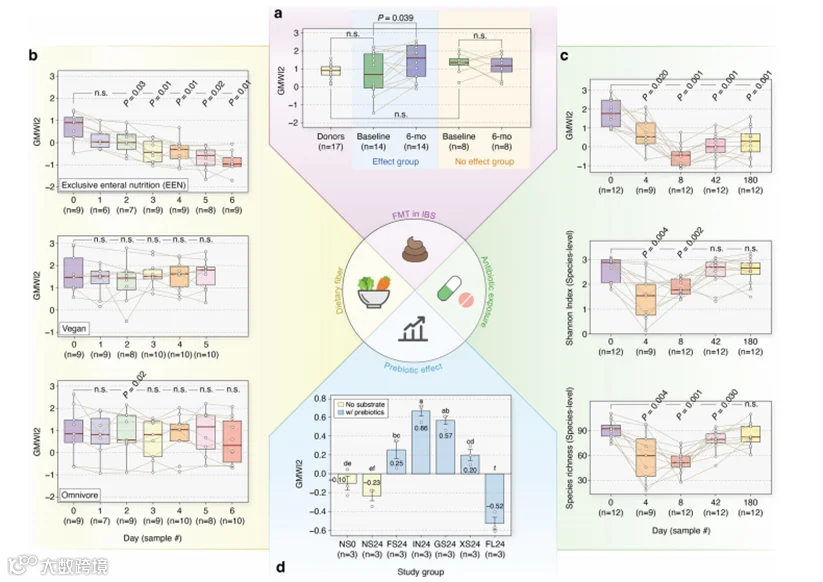
图6 | 使用GMWI2重新分析现有的纵向肠道微生物组研究。
讨论
最近对人类肠道微生物组的研究强调了其在预测性医疗保健工具开发中的潜力。在这方面,我们介绍了GMWI2,这是一个基于肠道微生物组分类档案的健康状况的强大预测器,与其原型(GMWI)相比,显示出显著的技术进步。我们广泛的多研究分析,汇总了来自54项已发表研究的8069个粪便宏基因组,涵盖了来自六大洲26个国家的多样化人群,以识别将肠道分类学与人类健康联系起来的生物信号。GMWI2为置信度较高的样本提供了约90%的交叉验证平衡准确率,确立了其作为区分健康和非健康表型的分类器的强大可靠性。此外,通过重新审视和重新解释先前发表的数据集,GMWI2甚至可以为饮食影响、抗生素暴露和FMT对肠道微生物组影响的既定理解提供新的视角。最后,本研究强调了广泛数据共享在促进稳健的机器学习应用以及展示对批次效应和偏见的抵抗力方面的重要性。
在我们的分析中,我们逐步增加了GMWI2的幅度截止值,我们认识到分类准确性和有资格进行类别预测的样本量之间存在一种反向关系。因此,将这个幅度截止值限制在单一值可能并不普遍适用;相反,这个参数的选择应该是灵活的,并由用户根据他们特定情境和个别数据集的可接受准确度阈值来决定。换句话说,用户可以根据他们对预测的信心水平偏好选择他们希望的GMWI2幅度截止值。这种用户驱动的方法,提供了在有限数据集中高度信心与更广泛范围预测但信心较低之间的灵活性,是我们的方法相对于传统二元输出机器学习技术的明显优势。此外,我们的发现因此促进了“拒绝选项”对于低GMWI2幅度的潜在效用,这可以作为将相对不确定的预测重定向到其他筛查方法的标准——这一概念捕捉到了健康和疾病的某些方面不能仅通过肠道微生物组完全解释的理解。
我们的研究虽然提供了对肠道微生物组预测能力的见解,但也有一些需要承认的局限性。首先,我们强调GMWI2分数反映了与健康状况的关联,我们根据疾病的存在或缺失来定义健康状况。重要的是要理解这些分数并不意味着与(也不打算取代)直接临床健康措施的因果关系,例如在胃肠道中检测致病生物、肠道运动特征、代谢剖面、血清学标记、血液炎症标记或粪便钙卫蛋白水平。其次,该模型可能受益于包括更复杂的微生物组特征,如物种生长速率、菌株细节和功能潜力。纳入这些重要因素可能会提高预测准确性,并提供关于将肠道微生物组与整体人类健康联系起来的复杂机制的更丰富视角。第三,我们努力确保我们汇总的粪便宏基因组数据集展现出地理、种族和文化的多样性代表。尽管如此,未来的工作应该强调更广泛的参与者包容性,特别是来自代表性不足的地区和种族,以真正全球化肠道微生物组研究。此外,放宽我们的选择标准将允许我们纳入更广泛疾病表型(如神经退行性和精神障碍)的宏基因组,并达到更多样化的人群。这种扩展可以增强模型在不同人群中的普遍适用性。第四,尽管我们使用了到物种水平的分类信息,但没有关注微生物菌株,这通常具有更多的临床意义,可能错过了一个机会。虽然我们的方法超越了16S rRNA基因扩增子测序的属级限制,但它并不考虑同一物种菌株之间的变异性。第五,我们的分析揭示了包括肠球菌在内的已知病原体在我们的GMWI2框架中没有显示出负系数。尽管如此,我们确实观察到某些机会性致病分类群的负系数,尤其是在各种梭菌物种中,如补充数据4中详细说明的。重要的是要强调,病原体特征的确定更准确地在菌株水平上进行,这超出了我们模型的范围。此外,广泛承认并非每个与慢性非传染性疾病相关的肠道微生物组都必然携带侵入性病原体。第六,我们认识到我们的模型通过可能受到通过时间、粪便一致性和其他未在我们的元数据中捕获的因素影响的健康和非健康之间的组成变化。这是个体样本的一个有效考虑。然而,在我们分析8000多个宏基因组样本时,我们的假设是,鉴于我们研究样本人群固有的广度和合理的多样性,这些变量可能均匀(随机)分布或对GMWI2工具的整体性能影响最小。最后,我们对健康(即自报无疾病或疾病相关症状)和非健康(即患有临床诊断疾病的患者)的定义与我们之前的研究中使用的定义一致,因为当前工作代表了我们之前方法的持续改进。然而,我们还没有研究这些定义的微妙变化如何影响GMWI2分类准确性。分析这一方面是未来研究的潜在领域。
关于其转化潜力,GMWI2旨在通过分析肠道微生物组分类档案,提供一种新的方法,以半实时方式动态监测个体的健康状况。虽然我们的指数明确训练用于区分健康和患病的肠道微生物组,它也提供了一种实用的方法来近似预测疾病状态。这是通过在健康和患病状态之间进行插值来实现的,允许GMWI2揭示肠道微生物组健康谱系上的变化。具体来说,假设我们的模型具有足够的预测质量,随着个体从健康状态转变为疾病前期再到疾病状态,他们的GMWI2分数将减少,或者如果向相反方向转变则增加。此外,GMWI2提供了一种实用的替代方案,用于资源密集型的收集纵向肠道微生物组数据集,这些数据集需要精确跟踪从健康到疾病的稳定过渡。目前这一领域的努力规模非常有限且成本高昂。
总的来说,GMWI2不是用来确认特定疾病诊断的,而是作为一个早期预警系统,类似于煤矿中的“金丝雀”。它的设计是为了在特定、可诊断的症状出现之前,检测整体肠道健康的潜在不利变化。这种检测可以通知饮食或生活方式的改变,以防止轻微问题升级为严重的健康状况,或促使进行进一步的诊断测试。与现有的特定疾病指数不同,我们的指数跨越了多种疾病,从而强调了一种泛疾病(或者更准确地说,一般健康)的肠道微生物组特征。这种广泛的适用性在临床场景中可能特别有用,例如在选择FMT捐赠者时,肠道健康可以被视为整体健康的反映。在类风湿性关节炎和其他自身免疫性炎症性疾病的情况下,GMWI2可以指导减少或停止治疗的决策,或评估疾病复发的可能性。从这个意义上说,GMWI2可能会开启一个以肠道微生物组为中心的健康分析的转型时代,允许进行针对个体微生物特征的细微健康评估。展望未来,将GMWI2整合到一个更大的决策网络中,与其他生物测量(例如,多组学、可穿戴设备)和AI模型一起,有可能为由肠道微生物组驱动的健康老龄化和预防性健康筛查以及健康计划带来激动人心的可能性。
往期推荐
