
奇辉观点
既往的研究多关注肠道微生物组的细菌成分与自闭症谱系障碍(ASD)之间的关联,ASD中肠道古菌、真菌和病毒,或肠道微生物组的功能是否发生了变化目前尚不清楚。与神经正常发育的儿童相比,ASD儿童显示出古菌、细菌和病毒多样性的减少,本文研究者对多王国和功能性微生物组进行了全面分析:ASD儿童中,有14种古菌、51种细菌、7种真菌、18种病毒、27个微生物基因和12条代谢途径发生了变化;其中基于31个多王国标记物的模型为ASD诊断实现了高预测价值,这些模型在年龄、性别和队列中的可复制性能突出了它们作为ASD诊断有前途工具的潜力,未来可用于ASD的非侵入性诊断。
论文ID
本文译自:Su Q , Wong OWH, Lu W, et al. Multikingdom and functional gut microbiota markers for autism spectrum disorder. Nature Microbiology, 2024 Jul 8. doi: 10.1038/s41564-024-01739-1.
发表杂志:Nature Microbiology
影响因子:20
通讯作者:Siew C. Ng
作者单位:The Chinese University of Hong Kong
引 言
自闭症谱系障碍(ASD)是一种异质性的神经发育障碍,特征为社交、认知和行为障碍。尽管ASD的成因尚不清楚,但人们认为它与遗传和环境因素之间复杂的相互作用有关。在过去十年中,肠道微生物组已被证明在调节肠脑轴方面发挥中心作用,通过调节神经免疫网络并与大脑直接通信,可能对ASD的发展有所贡献。临床前研究表明,患有ASD的儿童肠道微生物组成发生了变化,并且肠道微生物的发展延迟。此外,将来自ASD个体的粪便微生物群转移到无菌小鼠体内促进了类似自闭症的行为,而将健康个体的粪便微生物群移植到ASD儿童体内则导致症状的改善。到目前为止,大多数研究都集中在肠道微生物群的细菌成分上,并揭示了许多变化,尽管在ASD儿童的微生物多样性和组成上结果并不一致。宏基因组测序技术使得研究其他微生物群落成为可能,包括古菌、真菌和病毒,这些微生物也定植在人类肠道中,这些“暗物质”可能在ASD的发病机制中发挥关键作用。
在这项研究中,我们探索了对肠道古菌、细菌、真菌、病毒及其基因和功能的多王国分析,展示了1,627名被认为神经正常发育或患有ASD的儿童的宏基因组分析,这些儿童有广泛的表型数据,并在237份粪便宏基因组的公共数据集中验证了我们的发现。
结 果
研究特征
本研究共招募了来自五个独立队列的1,627名儿童(1-13岁,24.4%为女性)(图1a和扩展数据表1)。收集了包括年龄、性别、体重指数(BMI)、饮食、药物、共病、并发精神障碍、胃肠道(GI)症状(包括通过布里斯托粪便形态评分、BSFS评估的粪便一致性)、家庭特征以及与样本收集、储存和处理相关的技术因素在内的广泛表型数据(236个因素)。所有粪便样本均使用相同的标准化方案进行处理,以减少由技术因素引起的批次效应。我们总共获得了超过10TB的序列数据,每个宏基因组的平均深度为6.34吉基对(扩展数据图1)。在发现队列中,我们对709名ASD儿童和374名被认为神经正常发育的儿童(3-12岁,24.3%为女性)的粪便样本进行了宏基因组测序。在独立的医院队列中,我们对82名ASD儿童和90名被认为神经正常发育的儿童(4-11岁)的172份粪便样本进行了测序(图1a)。我们还包括了一个由较年轻儿童组成的社区队列,包括116名ASD儿童和60名被认为神经正常发育的儿童(1-8岁,29.5%为女性,图1a),以验证不同年龄组中的发现。共有237份粪便宏基因组(2-13岁,17.7%为女性)来自已发布的数据集,被纳入分析用于外部验证(图1a)。另外两个非ASD队列,包括注意缺陷多动障碍(ADHD,n=118)和特应性皮炎(n=78)的儿童,被用来评估我们发现的特异性(图1a)。
ASD相关的四个界微生物种类
由于肠道微生物组成的形成在很大程度上受到环境和宿主因素的影响,我们分析了236个宿主因素对肠道微生物组成的影响,以确定潜在的混杂因素(图1a)。在我们的发现队列中(3-12岁,n=1083,24.3%为女性),这些宿主因素的组合分别解释了古菌、细菌、真菌和病毒的个体间微生物组变异的12.5%、15.1%、10.7%和11.7%(图1b)。在所有研究的宿主和饮食因素中,共有21个因素对肠道微生物组成有显著影响,包括ASD、年龄、性别、BMI、3个GI参数、15个饮食因素和测序批次(扩展数据图2),因此这些因素在所有随后的分析中都进行了调整。接下来,我们评估了被认为神经正常发育的儿童与ASD儿童之间肠道微生物多样性的变化。与被认为神经正常发育的儿童相比,ASD儿童显示出古菌、细菌和病毒多样性的减少(图1c)。共有14种古菌、51种细菌、7种真菌和18种病毒在被认为神经正常发育的儿童和ASD儿童之间显示出不同的丰度(图1d)。在90种已识别的微生物种类中,有80种在ASD儿童中的相对丰度显著低于被认为神经正常发育的儿童(图1d)。这一发现在细菌群落中最为明显,ASD儿童中有50种细菌种类被耗尽,而只有一种细菌种类被富集(图1d)。ASD儿童中细菌种类的改变主要是由于Streptococcus thermophilus和产生短链脂肪酸的细菌(如Bacteroides sp. PHL2737和Lawsonibacter asaccharolyticus)的耗尽所驱动的。
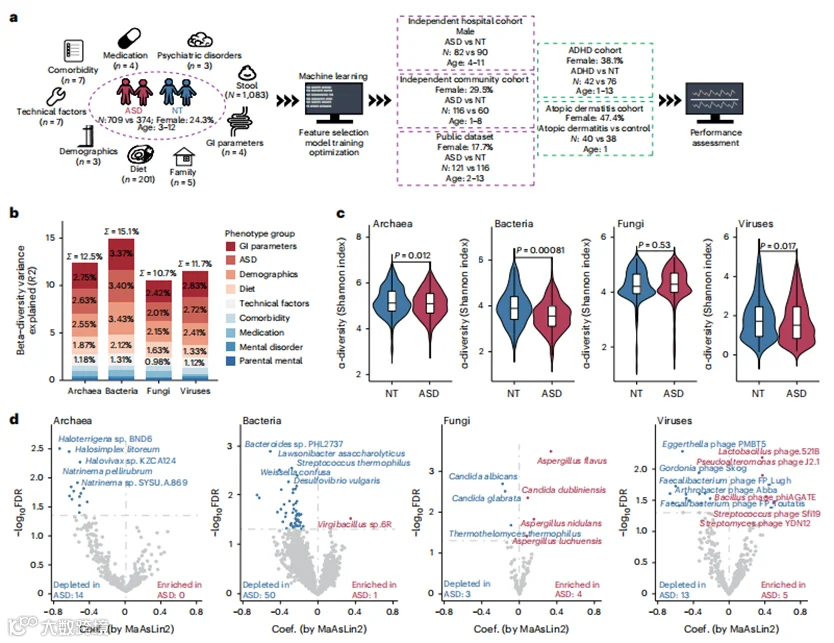
图1 | ASD与粪便微生物组组成的关联
a, 队列的图形摘要以及可用元数据的概览(n=收集的变量数量,N=样本大小)。b, 多变量PERMANOVA分析中,表型组解释的多王国(古菌、细菌、真菌和病毒)微生物组组成的变异。
c, 用Shannon指数测量的多王国(古菌、细菌、真菌和病毒)微生物组的α多样性,ASD儿童(红色,n=709)和被认为神经正常发育(NT,蓝色,n=374)的儿童。P值(双尾检验)是使用MMUPHin计算的。数据显示为箱形图,其中中位数(中心线),第25和第75百分位数(箱形图限制),以及第5和第95百分位数(须)。
d, 火山图显示了在调整显著混杂因素后,使用MaAsLin2计算的多王国(古菌、细菌、真菌和病毒)物种与ASD之间的关联。FDR<0.05的关联被认为是显著的,并分别用红色(ASD中富集)或蓝色(ASD中耗尽)标记。排名靠前的物种用名称标记。
自闭症肠道微生物组功能的转变
在功能层面,宿主表型因素分别解释了微生物组途径和微生物基因变异的17.1%和15.7%(图2a)。ASD的诊断是导致微生物组途径和微生物基因变异的首要因素(图2a)。共有19个宿主和饮食因素对肠道微生物组功能有显著影响,包括ASD、年龄、性别、BMI、2个GI参数、12个饮食因素和测序批次(扩展数据图3)。在调整了这些混杂因素后,我们鉴定了27个不同的京都百科全书基因和基因组(KEGG)同源性(KO)基因:与被认为神经正常发育的儿童相比,ASD儿童中有23个基因减少,4个基因增加(图2b)。在途径层面,我们注意到12条不同的途径,包括9条显示与ASD负相关的途径和3条显示正相关的途径(图2b)。我们发现,与神经正常发育的儿童相比,ASD儿童中辅酶Q7和磷酸硫胺素的生物合成途径减少(图2b)。辅酶Q具有抗氧化活性,并已被识别为能够改善ASD儿童症状的物质。磷酸硫胺素合成的损害与ASD和其他精神障碍在动物和人类的研究中有关。我们还观察到ASD与4-氨基丁酸(GABA)的绕路途径之间存在负相关。GABA是哺乳动物中枢神经系统的主要抑制性神经递质,并且在以前的研究中已与ASD相关联。
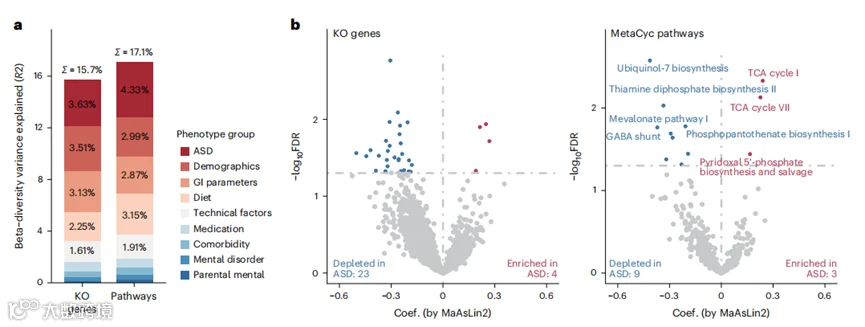
图2 | ASD与粪便微生物组功能之间的关联。
a, 在多变量PERMANOVA分析中,表型组解释的微生物组功能(微生物途径和基因)的变异。b, 火山图显示了在调整显著混杂因素后,使用MaAsLin2计算的微生物组功能与ASD之间的关联。FDR<0.05的关联被认为是显著的,并用红色(ASD中富集)或蓝色(ASD中耗尽)标记。排名靠前的途径用名称标记。
单一微生物界标记物在ASD诊断中的应用
已有几项研究探讨了用于ASD诊断的细菌标记物。然而,古菌、真菌、病毒、微生物基因(KO家族)或功能途径在ASD中的性能尚未被探索。为了避免因样本大小不平衡和残留混杂因素导致的歧视偏差,我们使用一对一配对算法,基于发现队列(n=1083,图3a),构建了一个ASD儿童(n=301,95名女孩和206名男孩)和神经正常发育儿童(n=301,95名女孩和206名男孩)的匹配子队列。配对后,在匹配子队列中,神经正常发育和ASD儿童之间的这些混杂因素没有差异(扩展数据图4)。这些结果表明,在神经正常发育或ASD的儿童中,环境和宿主背景是可比的,因此我们的模型不太可能受到这些重要因素的影响(图3a)。
在匹配的队列中,我们首先测试了使用单一王国标记物的模型来区分ASD儿童和神经正常发育的儿童的准确性。在所有单一王国标记物中,微生物途径模型显示出最强的预测能力,其平均曲线下面积(AUC)为0.87(图3b),其次是微生物基因(AUC 0.86)、细菌(AUC 0.85)、古菌(AUC 0.76)、真菌(AUC 0.74)和基于病毒的模型(AUC 0.68)(图3b)。总体而言,我们的发现表明,来自不同王国的粪便微生物组标记物为ASD诊断提供了有希望的预测能力。
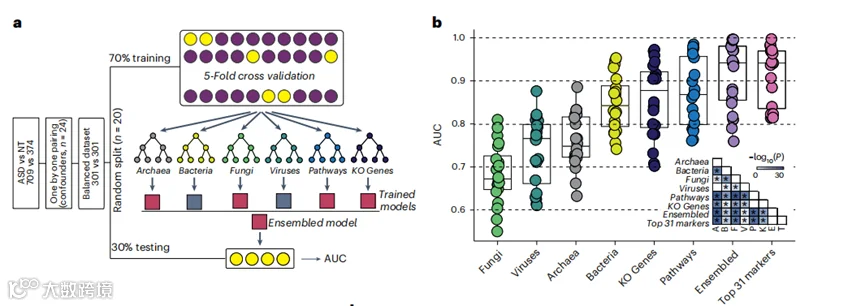
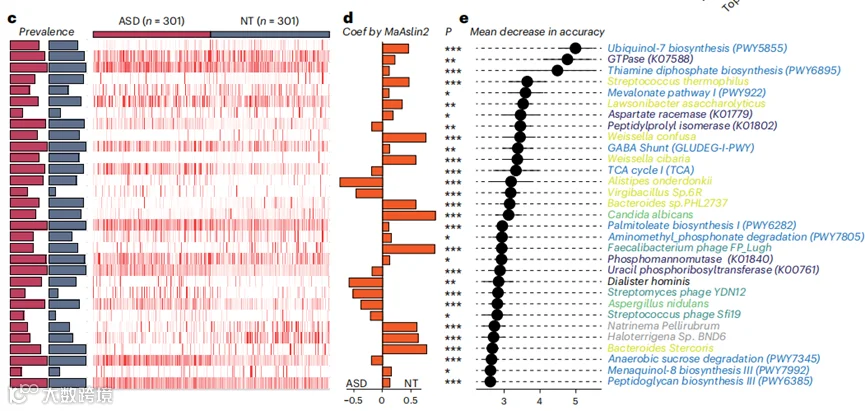
图3 | 用于ASD诊断的随机森林模型。
a, 随机森林模型发展的示意图。使用一对一配对算法从原始发现队列(n=1083)构建了ASD儿童(n=301)与神经正常发育(NT,n=301)的平衡数据集,考虑了24个显著混杂因素。采用了70%的训练和30%的测试样本分割,并使用测试数据来估计随机森林模型的准确性。这个过程随机重复了20次,以获得测试集上随机森林预测评估的分布。输入数据是一个包含六个组成部分的向量:古菌、细菌、真菌、病毒、微生物途径和KO基因。
b, 箱形图显示了20次模型训练和测试中得到的AUC得分的分布。使用双尾Wilcoxon秩和检验评估组间差异。数据显示为中位数(中心线)、第25和第75百分位数(箱形图限制)以及第5和第95百分位数(须)。
c, 在ASD儿童(n=301)和被认为神经正常发育(NT,n=301)的平衡数据集中,随机森林模型采用的前31个标记的流行率和相对丰度。
d, 使用MaAsLin2评估31个标记与ASD的关联(双尾检验,P值使用FDR方法调整)。*FDR<0.05,**FDR<0.01,***FDR<0.001。确切的FDR值提供为源数据。
e, 31个标记的准确性平均降低。
多王国微生物标记物在ASD诊断中的应用
接下来,我们探索了结合多王国特征的模型性能。我们发现,与基于单一王国特征的模型相比,集成模型在ASD诊断中表现出更优越的性能(平均AUC为0.91)(图3b)。这些结果证实了多王国粪便微生物组生物标记物小组在检测ASD方面的诊断性能高于单一王国小组。为了确定实现最高准确性的最小数量微生物组标记物,我们根据它们的排名,连续地将已识别的标记物纳入模型,最终,共有31个微生物特征在ASD诊断中显示出0.91的AUC(图3b)。这31个标记物的流行率和相对丰度在被认为神经正常发育的儿童和ASD儿童之间有显著差异(图3c)。基于MasAsLin2,21个标记物在ASD儿童中显著减少,而10个标记物显著增加(图3d)。我们重新分析了这31个特征的重要性,并观察到模型的准确性主要是由辅酶Q7生物合成途径、GTP酶和磷酸硫胺素生物合成途径驱动的(图3e),这支持了它们在ASD发病机制中的潜在作用。此外,包括Streptococcus thermophilus、Lawsonibacter asaccharolyticus、Weissella confuse、Weissella cibaria和Bacteroides sp. PHL2737在内的几种细菌的减少也是对诊断准确性贡献最大的微生物特征(图3e)。总体而言,我们的分析表明,一个包含31个粪便微生物组标记物的小组代表了一种有潜力的非侵入性工具,用于ASD的诊断。
在独立队列和公共数据集中的验证
为了外部验证诊断价值并避免对诊断准确性的过度乐观报告,我们在一个独立的医院队列中测试了这31个标记物的小组。我们发现,我们的模型保持了0.55到0.87的AUC范围(图4a)。其中,使用31个标记物的集成模型在AUC上排名第一,达到了0.87,敏感性为91%,特异性为73%(图4a)。这31个标记物中的28个在被认为神经正常发育和ASD儿童之间的相对丰度仍然显著不同(图4b)。此外,我们的模型在这个验证队列的一个较年轻子集(n=31,6岁或以下)中显示出0.61到0.89的AUC范围,其中使用31个标记物的集成模型再次实现了最高的准确性(扩展数据图5)。我们还测试了这个小组是否可以应用于预测另一个年轻队列(116名ASD儿童和60名神经正常发育儿童,1-8岁,29.5%为女性,扩展数据表2)中的ASD风险。训练好的模型达到了0.89的AUC,对男性和女性的表现相对平衡。当将年龄范围缩小到6岁或以下和4岁或以下时,模型显示出的AUC都是0.91。我们在这个按年龄分层的年轻队列中测试了这31个标记物与ASD之间的关联,并确认这些关联大多数仍然可以复制(图4b和扩展数据图6)。综合这些结果,证明我们训练的模型和31个标记物小组在年龄、性别和队列中的稳健性。
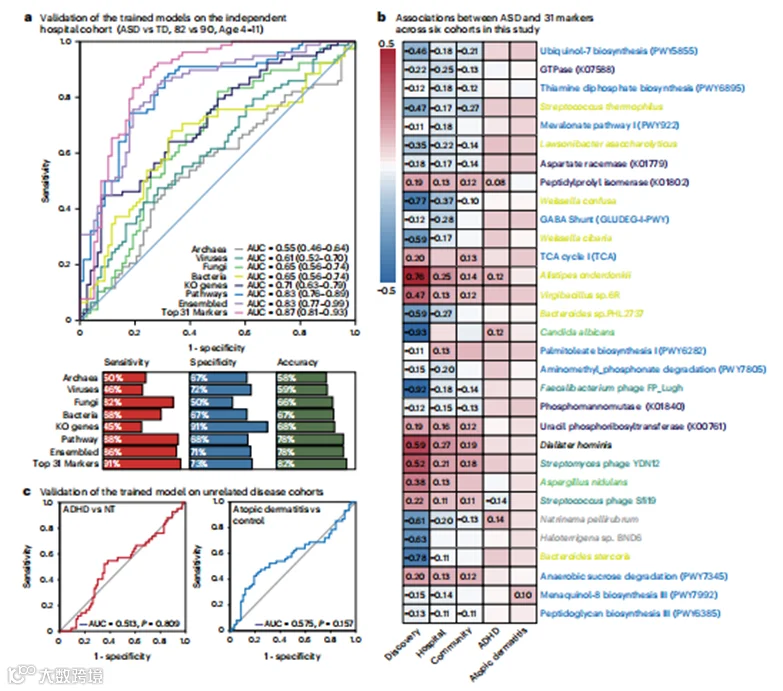
图4 | 随机森林模型的验证。
a, 在验证队列中使用不同特征的随机森林模型的AUC(95% CI)。敏感性、特异性和准确性是基于Youden指数计算的。在调整技术因素和包括年龄、性别、BMI、BSBF、功能性便秘和排便障碍在内的可用协变量后计算AUC。
b, 使用MaAsLin2(双尾检验)计算的五个队列中ASD与31个已识别粪便微生物组标记物之间的关联。仅当P<0.05时才标记Coef.值。c, 在独立的ADHD和特应性皮炎队列中测试使用31个标记的模型的AUC。P值是使用Wilcoxon秩和检验(双尾检验)计算的。
为了进一步测试我们的31个标记物多王国小组在不同人群中的可复制性,我们整合了来自亚洲、欧洲和美洲的六个公共数据集的237个宽范围粪便宏基因组数据(扩展数据图7a)。该小组在区分ASD儿童和被认为神经正常发育儿童方面显示出0.78的AUC,敏感性为65.30%,特异性为72.4%(扩展数据图7b)。更重要的是,它对男性和女性显示出相当的性能,证实了我们的模型适用于两种性别(扩展数据图7b)。总体而言,这个31个标记物的多王国小组可能在不同的人群和地理位置都相关。
多王国标记小组的特异性
考虑到不同疾病间共有的微生物组改变,验证我们确定的微生物生物标记小组的疾病特异性对于确保ASD诊断的低误报率非常重要。为此,我们在两个非ASD队列中评估了我们训练的模型,包括患有注意力缺陷多动障碍(ADHD,n=118)或特应性皮炎(n=78)的儿童(图1a)。ADHD和特应性皮炎被报道与肠道微生物组的改变有关。在我们的标记小组中,特应性皮炎或ADHD儿童的AUC值较低(ADHD的AUC为0.51,特应性皮炎的AUC为0.58;图4c)。此外,只有17名受试者被训练模型预测为ASD,反映出8.7%的低误报率。总体而言,这些结果支持31生物标记多王国小组对ASD的特异性。
ASD中辅酶Q7和磷酸硫胺素的减少
以往的研究表明,辅酶Q可以改善ASD儿童的症状。血浆中的硫胺(维生素B1)及其相关代谢产物,如磷酸硫胺素的浓度降低,被认为与ASD有关。然而,这些观察结果背后的原因仍然不清楚。我们发现,辅酶Q7生物合成途径和磷酸硫胺素生物合成途径的相对丰度主要推动了我们诊断模型的准确性,并且在三个队列中与被认为神经正常发育的儿童相比,ASD儿童中表现出一致的减少(图4b)。共有17种酶参与这两条途径,它们大多数在不同队列中的ASD儿童中被耗尽(扩展数据图8)。总的来说,这些发现强调了肠道微生物组中辅酶Q7和磷酸硫胺素生物合成基因丰度的减少似乎与ASD强烈相关。
讨 论
大多数研究主要集中于ASD中的肠道细菌变化。最近,研究揭示了非细菌微生物,如古菌、真菌和病毒在肠脑轴中的关键作用。然而,它们在ASD中的研究却很少。在这项研究中,我们使用来自5个独立队列的1600多个宏基因组,对多王国和功能性微生物组进行了全面分析。我们展示了古菌、真菌、病毒种类和功能性微生物组途径也能区分ASD儿童和被认为神经正常发育的儿童。我们证明了基于31个多王国标记物的模型为ASD诊断实现了高预测价值。这些模型在年龄、性别和队列中的可复制性能突出了它们作为ASD诊断有前途工具的潜力。
我们发现了一系列的细菌和非细菌标记物,并分析了它们与ASD的关联。其中,我们观察到几种有益细菌,如Streptococcus thermophilus、Weissella confusa和Weissella cibaria,它们与ASD呈现负相关。一些细菌标记物在先前的研究中也有报道,如Bacteroides stercoris。然而,很少有研究关注ASD与古菌、真菌和病毒之间的关联,因此这些我们报告的标记物需要在未来的研究中进一步探索。此外,我们发现特定的微生物功能可能通过干扰辅酶Q和磷酸硫胺素的生物合成而对ASD的发病机制有所贡献。辅酶Q和与硫胺相关的代谢产物在心理健康和神经信号传导中发挥着关键作用。我们的发现为肠道微生物组的磷酸硫胺素生物合成可能也是未来治疗靶点提供了进一步的证据。
尽管已有几项研究试图确定ASD的可复制微生物生物标记物,但在不同队列中的充分验证很少。研究之间缺乏一致性引发了一个问题,即迄今为止获得的微生物结果是否反映了队列之间的内在生物学差异、实验偏差或统计功效不足,无法排除有意义的比较。基于我们的ASD宏基因组数据集,我们系统评估了宿主变量和技术因素对肠道微生物组的影响,并在整个分析中充分调整了已识别的混杂因素。此外,采用的机器学习模型发展算法来自先前的研究,该研究表明,不匹配的宿主变量会显著高估人类疾病二元分类的AUC,因为不匹配的混杂因素将降低机器学习模型专注于疾病本身的能力,从而降低其在队列中的稳健性。此外,尽管对多数类进行下采样,然后将环境/技术因素作为额外的协变量加入混合模型的方法是传统的,但在临床上可能更具挑战性,因为需要收集粪便微生物组之外的数据,并且在现实生活设置中更难以生物学解释;因此,本研究采用了基于粪便微生物组的随机森林模型来实现ASD的预测。最后,我们展示了使用多王国和功能标记构建的模型比单一王国标记具有更高的诊断准确性,特别是添加的微生物途径在其他人类疾病中也被证明具有很高的诊断和预后价值。此外,基于31个标记物小组的模型在队列、年龄和性别中实现了非常高的ASD诊断预测价值,表现出稳健的性能。尽管ASD没有改变疾病的治疗方法,但早期识别疾病进行全面评估和启动训练已被证明可以带来更好的社交和行为结果。我们的模型具有应用于临床的有前途的潜力,值得进一步探索。
此外,31个标记物小组包括了几个古菌、真菌和病毒,突出了非细菌微生物作为ASD诊断生物标记物的关键作用。多王国物种之间的相关性也可能发展成为复杂的微生物群集,作为ASD发病机制中的生态驱动因素。然而,这些多王国关联的功能能力尚未被研究,值得进一步分析。
关于ASD相关的肠道菌群失调是否仅由饮食偏好驱动的问题一直存在激烈的争论。我们的结果表明,饮食对ASD儿童的肠道微生物组有影响。然而,在我们的分析中,即使在矫正了饮食因素后,ASD相关的微生物组改变,包括微生物多样性和组成,仍然存在。同时,为了避免我们的诊断模型被环境和宿主因素误导,我们基于一对一配对算法构建了一个完全匹配的训练队列,从而减少了混杂因素的影响,并可能解释了我们模型在不同队列中的稳健性。我们还包括了两种已知与肠道微生物组改变有关的常见儿童疾病的分析,特应性皮炎和ADHD,并证明了我们的31个标记物小组仍然特定于ASD诊断。
分析生活方式、种族和地点异质的儿童队列为研究与ASD相关的微生物组提供了一个独特的机会。通过结合多个可能具有较低普遍性的小型医院基础和社区队列,我们能够更好地代表ASD病例和对照组的谱系。然而,这项研究有一些局限性。
我们对肠道微生物组如何与饮食偏好、宿主免疫和GI以及ASD行为症状联系的理解在横断面研究中是有限的,因此当前数据限制了我们进行因果推断的能力。尽管我们没有分析基因型对微生物组的影响,但先前的研究已经确定了与ASD高风险相关的基因。需要对遗传标记与微生物组小组结合的前瞻性研究,以确定它们是否可以进一步提高诊断准确性,更早地预测ASD以预防疾病。此外,尽管先前的研究已经充分描述了匹配算法,它也可能将未知的偏差引入结果,因此对训练模型进行独立验证和测试是必要的。我们已经初步证明了我们的模型在不同队列和年龄中的稳健性,对包括ADHD和特应性皮炎在内的无关疾病敏感性低,但是,我们模型的前瞻性验证试验在整个神经发育状况、儿童常见身体疾病和地理位置的范围内将非常需要,以进一步证明这些标记的普遍性和临床适用性。最后,尽管我们的模型在不同年龄、性别和队列以及公共数据集中表现出良好的性能,但仍可能存在未测量的混杂因素或难以避免的批次效应的残余可能性,这可能导致性能过高,因此在临床应用前需要独立的第三方验证。
总之,这项研究提出了一个高度特异性的多王国微生物小组,用于ASD的非侵入性诊断。从结合分析异质性ASD形式中开发可复制的微生物组生物标记物和准确的疾病预测模型,为未来的临床诊断测试和假设驱动的机制研究奠定了基础。
往期推荐
