
真核细胞中染色质的基本单位是核小体,它是由 147 个碱基及缠绕其上的八聚体组蛋白构成,这些组蛋白的主要成分是 H2A、H2B、H3、H4,连接组蛋白 H1 与非组蛋白[异染色质蛋白 1(HP1)]会与核心组蛋白协同作用共同形成更为有序的染色质结构,即异染色质。这种更有序的结构对于 DNA 复制、转录、DNA 损伤修复都有调控作用。组蛋白修饰主要是通过对组蛋白的氨基酸进行修饰,包括组蛋白乙酰化、甲基化、磷酸化、泛素化等。这些修饰通过单独作用或相互协作以改变染色质的开放程度决定某些基因的表达。组蛋白修饰方式中与衰老关系最为密切的是组蛋白甲基化及乙酰化修饰。儿童早衰症与成人早衰症是基因组的无序而导致的人类早衰模型,这两种疾病的早衰表型与正常生理的表型非常的一致,是作为研究人类衰老进程的典型模型[1]。这两种疾病分别是由于核膜蛋白及 DNA 损伤修复蛋白突变,导致染色质结构改变而引起的。同时在人的间充质干细胞中进行 WRN 基因敲除后,发现这种 WRN 缺陷的细胞表现出早衰的表型,并且整体 H3K9me3 水平下调,并且 WRN 蛋白可以与异染色质蛋白 HP1α、SUV39H 相互作用,维持异染色质结构稳定性[2]。这些都暗示我们,基因组不稳定及染色质结构紊乱是衰老的重要诱因。
关于衰老的最初的假说是“异染色质丢失”理论。这个理论认为异染色质丢失导致整个细胞核结构都发生改变,从而定位在核内的基因表达都会受到直接或者间接的影响,从而导致衰老。在一些早衰的细胞模型中也观察到异染色质的丢失。异染色质导致的基因转录的改变从酵母到人类中都有被检测到,转录组的改变对于基因的表达调控有上调也有下调,对寿命有延长也有缩短。但是关于异染色质丢失理论存在着悖论。衰老过程中伴随着异染色质的丢失同时也有衰老相关异染色质聚集的形成。之前的理论认为,衰老相关异染色质形成是个体衰老与复制性衰老的区别,但是最近通过 HI-C 及 FAIRE 技术分析发现,衰老细胞的最终异染色质状态经过 2 步,首先是整体异染色质的丢失,之后是某些特定部位的常染色质发生凝集形成异染色质,最终这两个步骤共同完成衰老细胞中异染色质重新分配[3]。
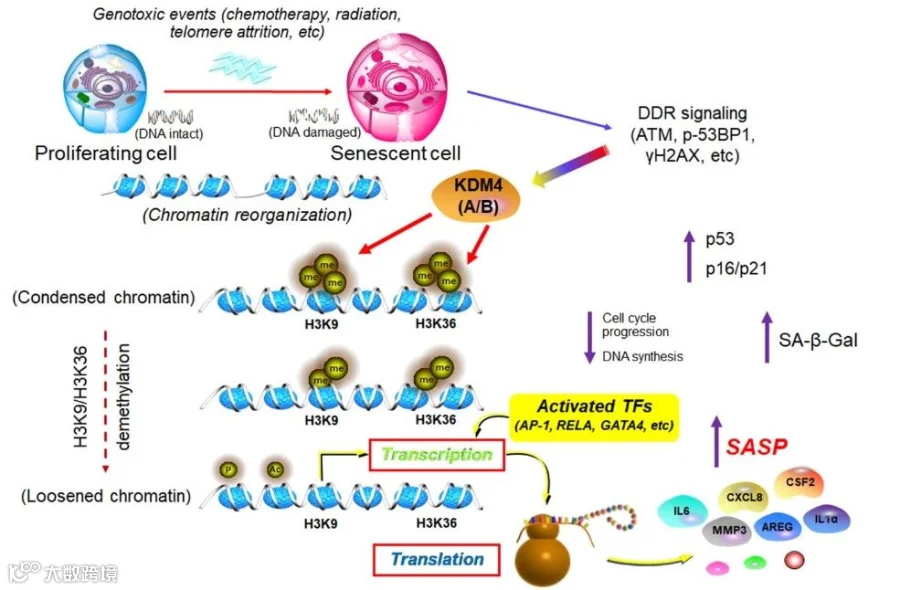
图1 组蛋白修饰调控衰老
组蛋白甲基化修饰
组蛋白的不同修饰方式会影响染色质的开放或者凝集,对基因表达的作用或是激活,或是沉默。组蛋白甲基化修饰一般是指在组蛋白赖氨酸残基上进行的甲基化翻译后修饰,受到组蛋白甲基转移酶调控。一般来说组蛋白 H3 第 4 位赖氨酸三甲基化(H3K4me3)与 H3 第 27 位赖氨酸三甲基化(H3K27me3)在转录调控中分别起到转录促进及转录抑制作用,在许多模型中展示出与寿命相关基因调控相关[4]。
衰老会引起这些激活与抑制的组蛋白修饰的不平衡,但是具体的机制研究有待挖掘。多梳蛋白(polycomb,PcG)和 Trithorax(TrxG)家族复合体是两类重要的组蛋白甲基转移酶复合体,它们通过对组蛋白赖氨酸残基进行特异性位点修饰并相互拮抗来实现对组蛋白的修饰。PcG 家族蛋白是成体干细胞重要的表观遗传调节因子,主要包含 PRC1 和 PRC2 两类蛋白复合体。PRC2 蛋白复合物主要包含 EED、SUZ12、RBAP46/48 及 EZH2/EZH1 四个成分,其中 EZH2/EZH1 具有甲基转移酶活性,可以特异性地催化组蛋白 H3第 27 位赖氨酸甲基化。TrxG 家族主要是属于 COMPASS 蛋白家族[5]。COMPASS 蛋白及其同源物在果蝇、小鼠以及人类中都起到对组蛋白 H3 第 4 位赖氨酸进行修饰的作用。PRC1 蛋白复合物中的具有泛素连接酶活性的 RING1A/B 蛋白,主要对组蛋白 H2A 第 119 位赖氨酸进行泛素化(H2AK119ub)修饰,同时 PRC1 复合物中其他成分也会对组蛋白 H3第 27 位赖氨酸进行甲基化修饰。组蛋白的甲基化修饰对于核小体的结构维持起到重要作用,从而对发育或衰老相关基因表达、DNA 修复等起调控作用。
在核膜蛋白 LaminA/C 基因突变的人成体干细胞早衰模型中发现组蛋白甲基转移酶 EZH2 较少,并且整体组蛋白第 9 位赖氨酸三甲基化及其结合蛋白 HP1α 水平降低,异染色质丢失[6]。此外,研究表明,Dot1/DOT1L 是特异性地使组蛋白 H3 第 79 位赖氨酸发生甲基化的组蛋白甲基转移酶,而 H3K79 甲基化与端粒区域的沉默、发育、细胞增殖检验点、DNA 修复、基因转录等相关[7]。
组蛋白乙酰化修饰
组蛋白乙酰化修饰指在组蛋白乙酰转移酶和去乙酰化酶催化作用下,对 H3、H4 的 N-端赖氨酸残基进行修饰。组蛋白乙酰化修饰通过疏松核小体结构,使其中转录因子特异性结合的 DNA 序列暴露出来,发挥激活基因转录的作用[8],报道指出,组蛋白乙酰化修饰参与寿命调控。在大脑以及肝脏等组织器官中组蛋白乙酰化水平的变化与衰老性组织退行密切相关。在裂殖酵母复制性衰老中,乙酰化修饰的主要方式是 H3K56Ac、H4K16Ac。报道指出,组蛋白 H3K56Ac 水平会影响染色质组装、基因组稳定性、DNA 复制、基因表达等生命过程。当去乙酰化酶 H3、Hs4 基因敲除后,H3K56Ac 的水平随之降低,酵母基因组不稳定性增加,最终导致酵母寿命缩短。与在衰老过程中 H3K56Ac 水平下调相反,H4K16Ac 水平上调,并且是通过影响端粒区的染色质结构实现对衰老的调控。敲除 H3 去乙酰化酶复合物 Rpd3 促进果蝇与酵母的寿命延长。在小鼠大脑中发现,DNA 重复元件上整体组蛋白呈现低乙酰化状态,并且年老的小鼠出现记忆缺失,这种记忆缺失被认为与促进转录延伸的 H4K12Ac 水平较低相关。当 H4K12Ac 水平恢复后,发现可以改善小鼠伴随衰老而出现的记忆缺陷现象[9]。
HDACsIII(sirtuin)家族蛋白与衰老密切相关。与酵母 Sir2 同源的人类 sirtuin 家族蛋白对维持衰老过程中成体干细胞的稳态至关重要。SIRT6 与 SIRT7 蛋白都属于 NAD+ 依赖的组蛋白去乙酰化酶,SIRT6/SIRT7 缺失造成小鼠早衰并引起寿命的缩短,提示 SIRT6/SIRT7 具有潜在的抗衰老作用。SIRT6 纯合性敲除的人间充质干细胞表现出加速衰老的特征。SIRT6 与 NRF2 相互作用并且使 H3K56 发生去乙酰化,从而招募 RNA 聚合酶Ⅱ复合物,发挥对 NRF2 靶基因的转录激活作用,SIRT6 纯合性敲除的人间充质干细胞表现出整体组蛋白 H3K56Ac 水平升高,无法招募 RNA 聚合酶Ⅱ结合到 NRF2 的启动子区域,细胞表现出衰老表型[10]。这项发现对于深入理解 SIRT6 对衰老和寿命的调控及探索衰老相关疾病的干预具有重要意义。另外体外实验也证明,SIRT1 敲低人视网膜干细胞出现早衰的表型,过表达 SIRT1 后,这种早衰的表型可以被挽救。此外,小鼠模型中发现,Cdc42 活性激活会抑制核膜蛋白 LaminA/C 蛋白表达,减少 H3K16Ac 的分布,影响染色体的状态,致使造血干细胞衰老[11,12]。
参考文献
扫码关注获得更多内容

小红书@一米生物

视频号@一米生物

知乎@一米生物
公司简介


微信号|一米生物
服务热线|400-097-3606
